



第四节 核苷酸代谢酶缺陷
目前已确定红细胞核苷酸代谢酶中有3种酶的病变可导致溶血性贫血,其中两种有鲜明特点,嘧啶5′-核苷酸酶缺陷以大量嗜碱性点彩为红细胞形态学特征,而腺苷脱氨酶为唯一的酶活力异常增高而非缺陷导致溶血性贫血的酶病。这类酶缺乏导致的溶血机制主要为核苷酸代谢池平衡紊乱,引起细胞中毒或缺乏能量而溶血。
一、嘧啶5′-核苷酸酶
【流行病学】
嘧啶5′-核苷酸酶(pyrimidine-5′-nucleotidase,P5′N,EC 3.1.3.5)缺陷不仅在核苷酸代谢酶中发生率最高,在所有导致溶血的红细胞酶病中发病率也占前,与葡萄糖磷酸异构酶(GPI)并列为第三位。1974年Valentine报道首例红细胞P5′N缺乏症,至今见刊60余例,世界不同地区均有报道,国内报道2例。鉴于病例数的增长情况,P5′N缺陷发病率有超过GPI的趋势。
【同工酶性质】
红细胞嘧啶5′核苷酸酶(P5′N)又称胞质5′核苷酸酶Ⅲ(cytosolic 5′-nucleotidaseⅢ,cNⅢ)或称尿苷一磷酸水解酶-Ⅰ(uridine monophosphate hydrolase type-1,UMPH-1),属于胞质5′核苷酸酶家族(CN),该家族是催化单磷酸核苷脱磷酸酶功能群组酶(EC 3.1.3.5和3.1.3.6),人类5-单磷酸核苷酶至少有7种,动力学性质和亚细胞定位各不相同。P5′N有两种形式同工酶:P5′NⅠ和P5′NⅡ,分别由不同基因(NT5C3和NT5C)编码,表现不同的分子量和动力学性质。P5′NⅠ主要在红细胞中高表达,P5′NⅡ则在机体广泛分布。P5′NⅠ对嘧啶类底物特异,对嘌呤类核苷酸完全无活性。P5′NⅠ既有一磷酸嘧啶核苷的磷酸水解酶作用,降解RNA来源的一磷酸嘧啶核苷(UMP、CMP),又具有嘧啶核苷酸磷酸转移酶的作用,在嘧啶核苷酸受体之间转移磷酸基团(图5-10)。P5′NⅡ只有磷酸水解酶作用而没有磷酸转移酶的功能,对嘌呤类和嘧啶类具有同样活力,尤其对DNA来源的一磷酸脱氧核苷(dUMP、dCMP)活力高。另一种机体广泛分布的胞质5′核苷酸酶Ⅱ(cNⅡ)主要以嘌呤核苷酸为底物,它也具有磷酸转移酶功能。转移磷酸基团的能力与磷酸与酶中间体较高的稳定性有关。在46℃条件下研究P5′NⅠ半衰期,显示为一个稳定的酶蛋白。P5′NⅠ活力最适pH在7.5~8.0,但是酶最稳定的pH在6.0~7.5。P5′NⅠ储存在pH 6.5、4℃条件一个月仍能保持原有活性,但是温度低于-20℃,活力将丧失40%。临床已利用这些核苷酸酶的磷酸转移酶功能,使用核苷酸类似物如叠氮脱氧胸苷和阿糖胞苷等,经体内代谢产生活化形式,治疗肿瘤和病毒性疾病。
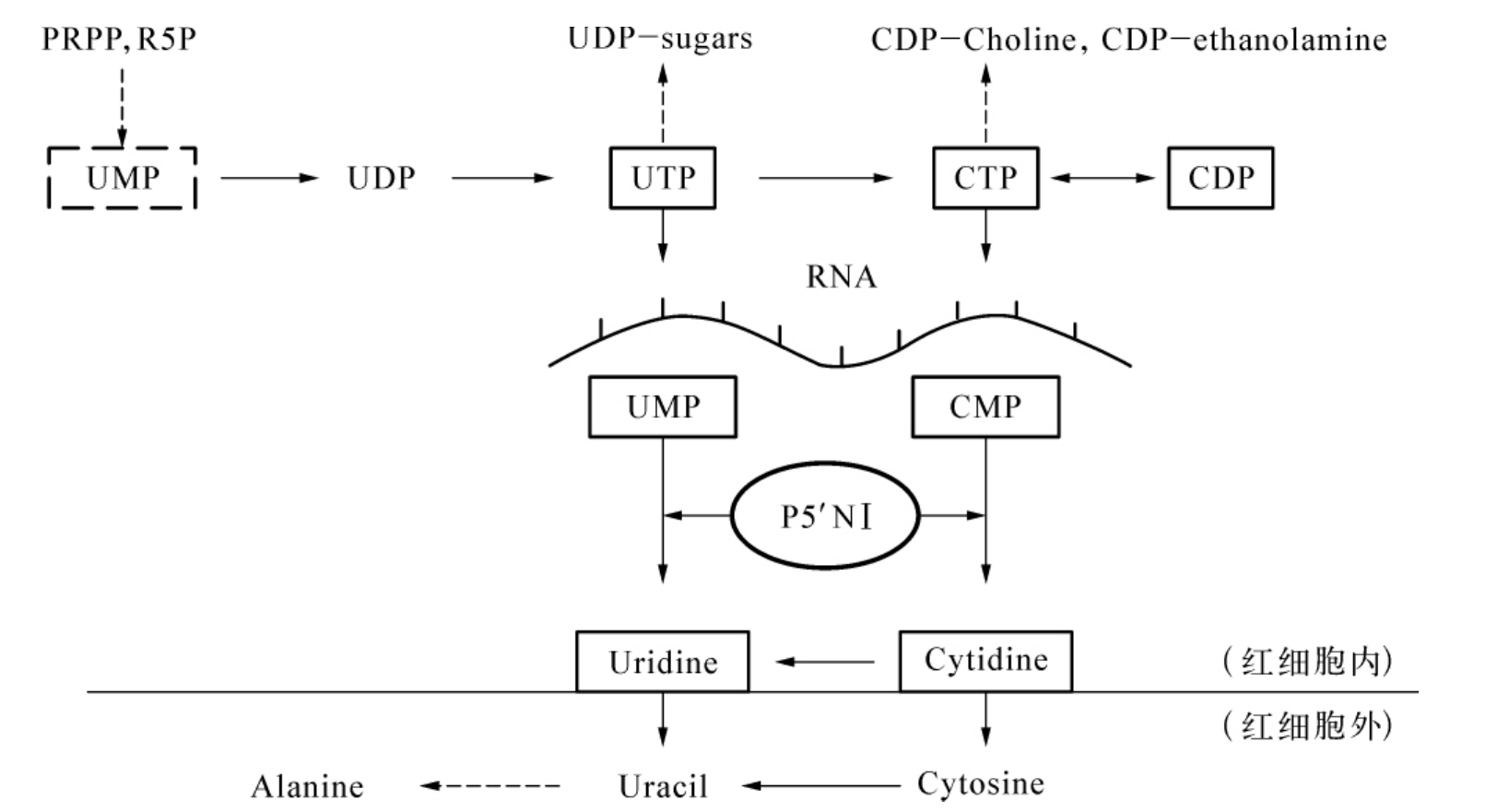
图5-10 P5′NⅠ催化途径及相关核苷酸代谢
PRPP:磷酸核糖焦磷酸;R5P:5′-磷酸核糖;UMP:一磷酸尿苷;UDP:二磷酸尿苷;UTP:三磷酸尿苷;Uridine:尿嘧啶核苷;Uracil:尿嘧啶;Alanine:丙氨酸;UDP-sugars:尿苷二磷酸糖类;CDPCholine:胞二磷胆碱;CDP-ethanolamine:胞二磷乙醇胺;CTP:三磷酸胞苷;CMP:一磷酸胞苷;Cytidine:胞嘧啶核苷;Cytosine:胞嘧啶
与缺陷率在遗传性红细胞酶病前两位的葡萄糖-6磷酸脱氢酶(G6PD)和丙酮酸激酶(PK)不同,P5′NⅠ至今未得到晶体蛋白,所以三级结构仍不明,远紫外圆二色性分析显示二级结构37%为α螺旋结构,26%为伸展的β链。质谱分析显示P5′NⅠ有3个分子量呈倍数关系的蛋白峰(26952、55476、110938),而P5′NⅠ在琼脂糖和聚丙烯酰胺凝胶电泳中仅显示单一区带,提示P5′NⅠ可能为一种多亚基组成的变构酶。
【P5′NⅠ功能及酶缺乏溶血机制】
红细胞中主要表达P5′NⅠ型同工酶,目前只观察到P5′NⅠ缺陷性溶血性贫血,未见P5′NⅡ相关疾病。P5′NⅠ的独特作用是在红细胞成熟过程中清除RNA的降解产物。网织红细胞及晚幼红细胞含有RNA,当红细胞成熟脱核脱网时,RNA被核糖核酸酶降解为各种核苷酸,其中腺嘌呤核苷酸(ATP、ADP、AMP等)可以被细胞再利用,而一磷酸嘧啶核苷(UMP、CMP)不能被红细胞利用,也不能透过红细胞膜,必须被水解成嘧啶核苷后才能排出细胞外。P5′NⅠ专一作用于嘧啶类5′-单核苷酸(CMP、UMP)的磷酸酯键,将其转变为可从胞内向外运出的核苷,从而维持细胞内核糖体、RNA及游离核苷酸代谢平衡。由此可见,P5′NⅠ对网织红细胞阶段的代谢特别重要。红细胞P5′NⅠ缺陷可使嘧啶核苷酸脱磷酸反应减弱,导致网织红细胞脱网障碍、核酸代谢紊乱,细胞内大量嘧啶核苷酸蓄积使细胞中毒,进而影响红细胞功能,最终引起以外周血大量嗜碱性点彩红细胞为特征的溶血性贫血。
与其他年龄细胞群相比,P5′NⅠ在网织红细胞中活性特别高,增高幅度较之其他年龄依赖性红细胞酶都要明显,所以可作为年轻红细胞群的敏感标志物。根据P5′N的底物特异性和网红高活性,推测P5′N缺陷主要影响年轻红细胞的代谢,患者脾切除术后网织红细胞计数往往高于术前,可能也与此有关。
P5′NⅠ活性高度依赖于镁离子(Mg2+)存在,锰离子(Mn2+)不能替代Mg2+,金属螯合剂如EDTA可明显抑制酶活力。P5′NⅠ也易受重金属(Cd2+、Cu2+、Hg2+、Ni 2+、Pb2+等)、巯基反应试剂(如氯汞苯甲酸、N-乙基-马来酰胺、二硫代双硝基苯甲酸)抑制,所以重金属中毒可以导致获得性的P5′NⅠ缺乏,产生类似于遗传性P5′N缺陷溶血症状。
P5′N缺乏者的残余酶活力与溶血程度无明显相关性,可能其他核苷酸酶或其他核苷酸代谢途径可以补偿P5′N缺陷。P5′NⅠ缺乏时,嘧啶核苷酸蓄积对红细胞代谢的确切损伤机理仍不明,可能有多种作用途径:①增加嘧啶核苷单磷酸激酶活性,因为三磷酸胞苷(CTP)和三磷酸尿苷(UTP)与一磷酸胞苷(CMP)的比例不平衡。而且,CTP蓄积又可导致二磷酸胞苷(CDP)结合物(CDP-胆碱、CDP-乙醇胺)显著增加(图5-10),CDP-胆碱和CDP-乙醇胺都是红细胞膜的重要成分,而不平衡的异常膜磷脂组成可能是引起溶血的原因之一,因为尽管大多数患者红细胞渗透脆性正常,但仍有少数患者表现出红细胞渗透脆性增高。②嘧啶核苷酸也是已知的镁离子(Mg2+)的强力螯合剂,可能因过多蓄积而影响需要Mg2+做辅因子或Mg-ATP做底物的那些酶的活力。在P5′NⅠ缺陷红细胞中磷酸核糖焦磷酸(PRPP)合成酶活力下降,嘧啶核苷酸蓄积扣留Mg2+可能是部分原因,由此也影响依赖PRPP的腺嘌呤补救途径代谢,进一步影响核苷酸池的平衡。③蓄积的嘧啶核苷酸通过与ATP/ADP竞争糖酵解酶(PFK、PK等)上的结合位点,干扰(不是阻断)糖酵解通量,影响细胞供能。④有些P5′NⅠ缺乏患者G6PD活力较低,但还原型GSH升高,可能由于嘧啶类蓄积降低细胞内pH,损伤磷酸戊糖旁路代谢,并抑制氧化型GSSG向外运输。
【酶分子病变机制】
在高发生率的红细胞酶病中,P5′NⅠ的基因定位和基因结构最晚获得,酶蛋白三级结构至今仍不清楚,在此之前,酶动力学性质对鉴别变异型十分重要,有缺陷的P5′NⅠ常表现对特异底物结合力减弱(Km增高),热稳定性增高或者下降均有报道,酶反应的最适pH也可发生变化,而酶的电泳行为改变是最直观的鉴定指标,国外报道有P5′N电泳迁移率减慢或不改变,国内报道一种新变异型(P5′N-Shanghai型),表现电泳快迁移变化。
2001年查明P5′N的基因定位,2005年应用基因工程手段反转获得特定突变酶蛋白,对P5′NⅠ的生化性质有了进一步的阐述。P5′NⅠ的基因(NT5C3,UMPH-1)定位于7号染色体(7p15-p14),含有11个外显子(exon 1,2,R,3-10),通过改变exon 2和exon R的剪接,编码3个不同的mRNA,产物分别为285、286和297个氨基酸。红细胞中只分离出286氨基酸残基的P5′NⅠ,单体相对分子质量为34 000,起始位点在exon 1的3′-端,从exon 3开始延伸,外显子2、R被剪接除去。含有exon 2或既有exon 2又有exon R的转录本仅在网织红细胞中表达,不同mRNA差别的生物学意义尚不清楚。
已从至少30个P5′N缺陷家系鉴定出至少20种突变,突变多发生在外显子8和外显子9上,除了6个错义突变(A260T、T392C、G469C、A536G、G688C和T740C),其他为无义、缺失、插入、剪接位点改变。突变导致酶催化活性损伤或热稳定性下降,其中以酶的热稳定性下降占多数。
基因分析结果进一步证明,遗传性P5′N缺乏者残余酶活力与溶血程度无明显关联。纯合子L131P(T392C)和G230R(G688C)酶活力很低,但表现中度贫血,提示溶血代偿性良好,可能对其他核苷酸酶或其他代谢途径进行了补偿。所以,核苷酸酶活性作为酶病患者预后指标可能不恰当。
有些基因缺陷可能与地域性分布有关,如543delG在3名意大利南部地区患者发现,在5个无关家系但是都来自日本九州岛的患者中均有错义突变G688C,而插入突变710~711insGG的报道来自地中海沿岸的意大利和土耳其,在同一个核苷酸位点上插入突变(Alu重复序列330bp)还见于两名无关联的葡萄牙患者。在一个挪威家系显示密码子81 TAC-TAT多态性与P5′NⅠ突变连锁,在英国群体普查结果TAT等位基因频率为0.29。
P5′NⅠcDNA克隆成功有利于进行基因结构分析,在分子水平进行更精确的诊断,排查因铅中毒、珠蛋白生成障碍性贫血、血红蛋白E等疾病引起的继发性P5′NⅠ缺陷。但是,DNA分析还存在一些复杂因素,因为染色体4号和7号上还存在P5′NⅠ拟基因(pseudogenes)。
【临床表现】
目前所称P5′N缺陷即指P5′NⅠ缺陷,为常染色体隐性遗传疾病,杂合子无临床症状,纯合子可表现溶血性贫血。诊断时年龄3个月至64岁均有报道,可有新生儿黄疸史。患者P5′NⅠ活力与溶血程度没有相关性,通常表现轻至中度的长期慢性贫血,约15%的患者表现重度贫血。在感染、怀孕等情况可加重贫血,有时需要输血。
并发症中脾肿大普遍存在,50%的文献患者在明确诊断前已经切脾,近半数病例有胆结石。少数病例有智力障碍和生长发育迟缓,cDNA文库显示脑中有P5′NⅠ分布,但是脑中P5N活力难以评价,而且患者有半数来自近亲婚配家庭,患儿近亲家庭可能另有致病原因,所以有学者对智力障碍是否为并发症仍有疑义。少见并发症包括细小病毒B19感染所致再障危象、慢性踝部溃疡。铁过载是慢性溶血病程发展的可预测的并发症,在P5′N缺陷者也常见,合并血色素基因(HFE)突变者(如HFE C282Y纯合子和HFE H63D杂合子),即使从未输血也呈现铁过载。P5′N缺陷引起的溶血主要为血管外(单核-巨噬细胞系统),在1例报告有血管内溶血、尿路丢失铁、肾脏铁沉积的患者,P5′N缺陷特征不明显,血管内溶血者可继发缺铁。P5′N缺陷可与血红蛋白病(HbE、HbD-Punjab等)和红细胞膜病(spectrin缺乏等)并存,基因型为复合型双杂合子。
【诊断】
(一)临床诊断
1.符合CNSHA诊断 有不同程度的慢性贫血、脾大和黄疸,轻至中度贫血多见。网织红细胞升高,与血红蛋白水平无相关性,极少数患者可无贫血但有高网红、高胆红素。在未切脾者网红中位数为8%,在已切脾者大多数显示更高,为15%,这一现象在PK缺乏症切脾术后也可观察到,可能是脾脏选择性扣留酶缺陷网织红细胞。
2.病史与家族史 可有新生儿黄疸史。家族中通常双亲无症状,而血缘同胞可能有同类症状表现。注意询问是否存在近亲婚配。
3.并发症 胆石症、机体铁过载、下肢溃疡等为CNSHA常见并发症。低龄就诊患者中可见智力低下、发育障碍,包括学习语言与独立行走迟滞。
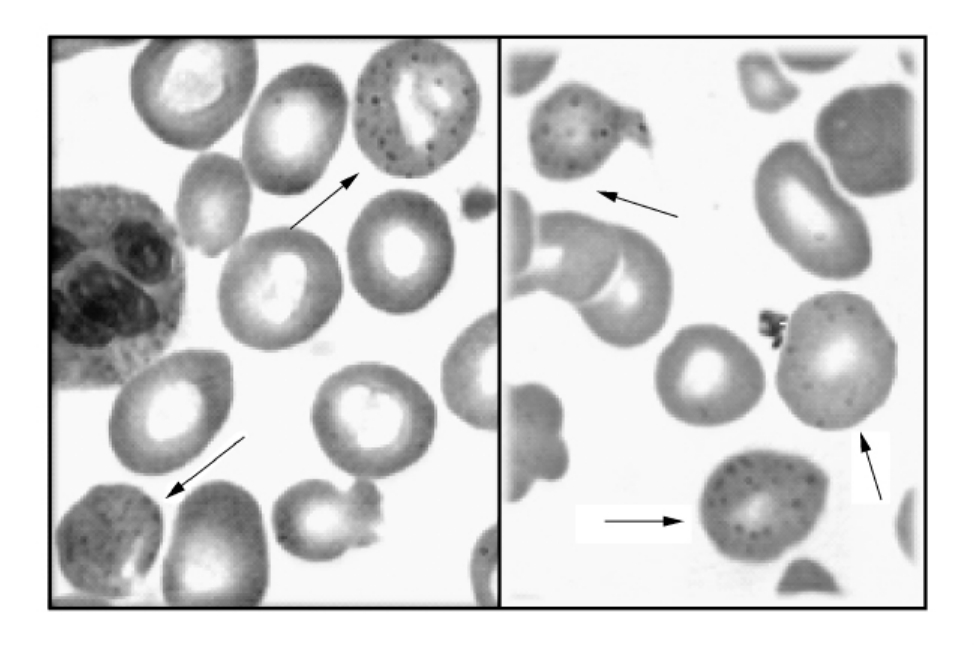
图5-11 嗜碱性点彩红细胞
(二)实验室诊断
1.红细胞形态 P5′N缺乏的突出特点是外周血涂片中红细胞嗜碱性点彩明显增多(图5-11),为2%~12%(正常值<0.3%)。多数患者的红细胞除了嗜碱性点彩增高以外,无典型的形态改变,少数患者血涂片可见球形、棘形红细胞。个别患者红细胞渗透脆性增高。
2.红细胞核苷酸光谱吸收比值(R260nm/280nm) P5′NⅠ缺乏的初筛试验。以嘌呤核苷酸(最大吸收峰在260nm)与嘧啶核苷酸(最大吸收峰在280nm)的光谱吸收值之比间接反映P5′NⅠ活力,正常值在2.29以上。P5′NⅠ缺乏时,嘧啶核苷酸蓄积,可使该比值小于2.29。
测定核苷酸的种类和含量还有纸层析法、毛细管电泳法、放射性同位素法、高压液相法等,其中毛细管电泳法重复性良好,有望应用于临床常规检查,但是需配备特殊实验仪器。
3.P5′N活力测定 P5′NⅠ缺乏的诊断依据。患者P5′N残余酶活力多在50%以下(1%~64%)。残余酶活力与溶血程度无相关性,与网织红细胞数量也无直接关系,支持嘧啶蓄积与代谢损伤主要发生在年轻红细胞的推测。
4.P5′N变异型鉴定 对确诊病例可按ICSH推荐方法进一步进行生化变异型分析,分析指标包括Km、Vmax、同类底物利用率、热稳定性、最适pH、电泳迁移率等。分子生物学方法用于鉴定7号染色体上P5′NⅠ基因突变型和基因多态性,应注意排除染色体4号和7号上P5′NⅠ拟基因的干扰。
【鉴别诊断】
(1)大量嗜碱性点彩是P5′N缺陷的明显病理表现,但是并非特异,其他一些遗传或获得性疾病也会出现嗜碱性点彩增多,如β珠蛋白生成障碍性贫血、某些血红蛋白变异型、铁粒幼细胞贫血、罕见的肝豆状核变性(Wilson’s disease)及铅、铜等重金属中毒,这些患者其他P5′N缺陷相关指标也相应改变。可用针对血红蛋白病的系列试验排除地中海贫血。铁粒幼细胞贫血可用骨髓细胞形态学和其他溶血指标予以鉴别区分。铅中毒多为特殊行业的成年患者,齿龈有沉淀线,血液和尿液的铅浓度超标,可用以鉴别。
(2)在有儿童期短暂幼红细胞减少症的患者,由于老年细胞群P5′N活力很低,易误诊为遗传性P5′N缺陷。可随访复查酶活力。
(3)血样放置后核苷酸吸收光谱会明显改变,出现假阳性结果。酶活力测定血样中原有核苷酸、游离磷酸物等杂质去除不尽会掩盖P5′N缺陷,所以实验诊断中应注意严密操作程序。
【治疗】
对P5′N缺乏溶血性贫血无特殊有效疗法。
(1)支持疗法为主。在感染、妊娠等因素加重贫血时,需要暂时输血治疗。一般情况下,真正依赖输血者很少。
(2)脾切除国外报道疗效不一,国内病例明显改善临床症状。文献病例统计,切脾可使Hb稳定在较高水平,术后Hb升高中位数32g/L(5~52g/L),对长期输血治疗者,术后无需再输血。但也有一些病例疗效不明显。
(3)适当补充叶酸,预防溶血危象或再障危象。铁过载者需用铁螯合剂。
二、腺苷脱氨酶
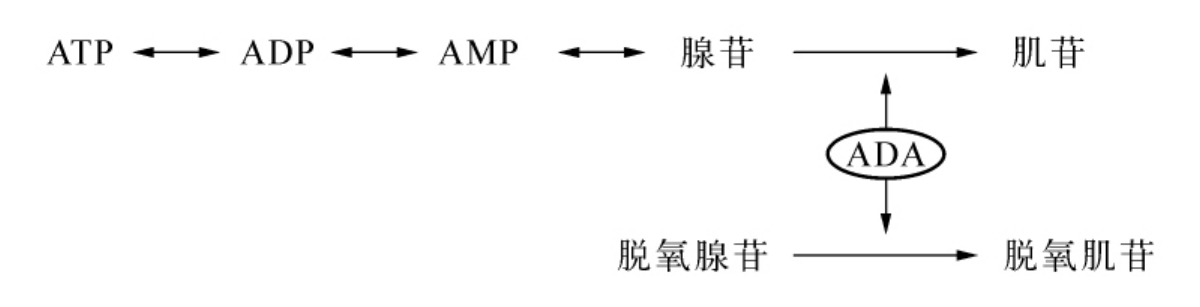
腺苷脱氨酶(adenosine deaminase,ADA,EC 3.5.4.4)催化不可逆的脱氨反应,使腺苷转化为肌苷(次黄嘌呤核苷)、脱氧腺苷转化为脱氧肌苷。ADA在不同组织中、不同发育状态酶活力可差别1 000倍,表达最高的组织细胞是皮质胸腺细胞和T淋巴细胞。有两种类型ADA遗传缺陷,产生截然不同的临床症状。一种是常染色体隐性遗传疾病,ADA的基因发生突变,导致ADA活力丧失、T淋巴细胞和B淋巴细胞耗竭,产生重症联合免疫缺陷病。另一种是常染色体显性遗传疾病,ADA的基因未见突变,ADA活力异常增高,导致溶血性贫血临床症状。
ADA是红细胞酶病中唯一的由于酶活力异常增高而引起溶血性贫血的酶,称为ADA过剩症(ADA overproduction),酶活力比正常对照高40倍以上,最高可达110倍。患者可出现程度不同的贫血、黄疸和脾脏肿大,网织红细胞计数增高,胞质ATP含量下降(与高网红对照相比,ATP含量下降至64%左右),红细胞寿命缩短。目前已报告20多个ADA过剩症家系。
ADA过剩症为常染色体显性遗传,基因定位于20号染色体。由ADA cDNA推论得到其酶蛋白序列362个氨基酸,相对分子质量为40 638。基因长度为32 000碱基对,含12个外显子。有报告显示人体存在两种ADA:低相对分子质量ADA(约40 000)主要存在于红细胞和淋巴细胞;高相对分子质量ADA(100 000~300 000)存在于其他组织。在溶血性贫血患者的红细胞中ADA活力异常升高,但是白细胞、成纤维细胞中ADA活力正常。
ADA导致溶血的分子机制不明。绝大多数红细胞酶变异是由于酶的结构基因突变所致,而ADA研究结果显示,ADA的基因序列、5′-区、第一内含子调节区、启动子均未见异常,ADA mRNA长度和序列也未见异常,ADA酶蛋白的代谢动力学性质正常。与正常对照不同的是,患者网织红细胞ADA mRNA含量明显高于正常人,而B淋巴母细胞ADA mRNA含量正常。对位于ADA的基因上游1.1kb的Alu元件(Alu variable poly A,Alu VpA)3′端高度多态性的重复序列尾部TAAA进行分析,结果一家系19名成员中,11名患者均有“12TAAA”重复序列,而8名正常者只有11TAAA以下的重复序列。这些研究结果提示,ADA过剩症发病机制可能为表达调控异常,在ADA的基因附近发生顺式活性突变使下调机制缺损,换言之,与正常ADA转录元件作用的反式调节因子可能发生突变,导致正常结构的ADA基因转录增加,或者是转录后加工过程发生改变导致RNA转录物稳定性增高,使得ADA酶蛋白合成过剩。ADA活性异常增高,过多地消耗底物腺苷,结果由腺苷合成的腺嘌呤核苷酸减少,干扰核苷酸池平衡,使ATP生成减少,导致红细胞供能障碍而溶血。
三、腺苷酸激酶
Mg2++ATP+───AMP←AKMg2++ADP +→─ADP
腺苷酸激酶(adenylate kinase,AK,EC 2.7.4.3)又名ATP-AMP磷酸转移酶(ATP-AMP phosphotransferase),为分布广泛的球形单体胞质酶,催化腺嘌呤核苷酸(ATP、ADP、AMP)之间可逆的高能磷酰基转移反应,活力依赖于Mg2+或Mn2+。AK主要功能是处理细胞能量利用的代谢信号、为核酸合成提供核苷酸,从而维持细胞内腺嘌呤核苷酸池平衡和能量内环境平衡。红细胞AK缺乏将使这一平衡产生紊乱而干扰细胞功能与寿命,导致溶血性贫血。
AK至少有6种同工酶,脊椎动物有其中4种(AK1~3和5),各种同工酶有不同的组织和亚细胞分布、不同的底物特异性。AK1为单体胞质酶,存在于红细胞、骨骼肌、心、脑等多种组织细胞;AK2存在于肝、肾、脾、心的线粒体内膜;AK3存在于肝和心的线粒体基质。AK1和AK3的基因定位于9号染色体的不同区域,AK2的基因定位于1号染色体长臂。目前鉴定的AK缺乏所致溶血性贫血均为AK1缺乏。AK1的基因位于染色体9q34.1-q34.3,约12kb长度,含有7个外显子,编码蛋白为194氨基酸残基,相对分子质量约为21 500。最近报道该基因可以被p53诱导产生另外一种产物AK1b,为膜绑结合形式,提示可能与依赖p53的细胞周期终止有关。
1969年报道首例AK缺乏溶血性贫血,为常染色体隐性遗传病,比较罕见,目前报道12个家系。AK缺乏症绝大多数为中至重度溶血,有黄疸、脾大、网织红细胞增高等CNSHA症状,部分患者有新生儿黄疸。患者红细胞渗透脆性、直接抗人球蛋白试验、异常血红蛋白、不稳定血红蛋白试验均无异常。除了AK活力下降,其他红细胞酶活力正常或升高,红细胞寿命缩短。有些患者血涂片可见有少量椭圆形、球形、口形和裂红细胞,均不超过6%。
AK缺乏症是多系统疾病,除了溶血性贫血,半数病例并发智力障碍、运动障碍等。智力和意识运动损伤与患者是否为近亲婚生无必然关联。另有AK缺乏并存PK或G6PD缺乏的报道。
AK纯合子残余酶活力一般低于正常值的50%,有些甚至测不出酶活力,醋酸纤维膜电泳也检测不出AK区带[如基因突变型A164G(Tyr→Cys)]。但是,对患者AK1病变机制研究显示,AK活性不能作为预后指标,因为贫血程度主要与酶分子突变类型有关而不是酶活性。例如,一对黑人亲姐妹酶活力同样低下,但是只有一人发病,另一人无临床症状,提示临床症状差异可能还有其他遗传背景等影响因素。又如AK的基因突变型107CGA→TGA(Arg→stop),产生含107个氨基酸的截短的酶蛋白(正常酶为194个氨基酸),患者AK活力测不出,但是溶血程度为轻度,伴有神经运动损伤。
纯合型突变可发生在近亲婚生患者,如nt 498~500或nt 501~503缺失突变,丢失140Asp或141Asp。AK缺乏也可遗传自父母两种不同的AK突变,如1例西班牙籍男婴为复合两种AK基因突变的双杂合子,G118A(Gly40Arg)并存G190A(Gly64Arg),有严重的新生儿溶血、脾大,需定期输血。
涉及酶活性中心和空间构象的突变与酶活力丧失和酶稳定性下降密切相关。正常AK1多肽链折叠为两个球状区域,球状区之间的界面上有底物结合位点。发生催化反应时,酶分子进行大幅度运动,形成开放型酶,可以结合底物MgATP和AMP,结合之后转变为封闭型酶,遮蔽活性中心与水的接触,以防ATP被水解。Gly64Arg突变发生在酶分子球状区域界面,减弱酶分子活动度,影响开放型结构成为封闭型结构,造成位阻而抑制AMP结合,使酶活力下降。目前所鉴定的AK1突变中,4种突变[G118A(Gly40Arg)、G190A(Gly64Arg)、C382T(Arg128Trp)、418~420delGAC(Asp140del)]使酶活力明显下降,138delG(移码突变,在91密码子提前终止)和C319T(R107stop)突变使酶活力完全丧失,而突变型A491G(Tyr164Cys)使酶蛋白稳定性明显下降。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。












