



第二节 磷酸戊糖旁路代谢及相关氧化还原代谢酶缺陷
这一类酶的缺陷均与红细胞氧化代谢障碍有关,虽然各个酶参与的代谢途径不同,但互相之间中间代谢产物密切互换,相互影响代谢进程,共同的特点是红细胞易受氧化损伤,氧化性药物可以诱发溶血及其他病症或者加重病情。
一、葡萄糖-6-磷酸脱氢酶
【流行病学】
葡萄糖-6磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G6PD,EC 1.1.1.49)缺乏症是最早确定的与溶血性贫血有关的遗传性红细胞酶病,也是发病率最高的酶病,在遗传性酶病中发病率居首位,是先天性溶血性贫血的主要原因之一。20世纪初意大利南部地区的医生观察到食用蚕豆可以导致急性溶血,将其命名为蚕豆病,1956年Carson首次明确报道蚕豆病由红细胞G6PD缺陷引起,定名为G6PD缺乏症,至今全世界罹患者已超过4亿人,被WHO定义为“与人类健康关系最大的高发遗传病之一”,全球发病人数居所有遗传病之首。
G6PD缺乏症地理分布广泛,种族间发病率差异很大。通过大量群体普查和对不同地区与民族的变异型分析,发现从北非、地中海、中东到东南亚这些地处北纬10°~35°的地区形成一条高发病地带,在非洲某些地区达35%,在土耳其东南部的犹太人中发病率可达58.2%。我国的发病率为4%~15%,华南、西南地区为高发区,包括海南、广东、广西、云南、贵州、四川等省,而黄河流域及黄河以北地区发病率较低,如广西某些地区为15.7%,云南傣族为16.6%,而河南杞县为2.53%,宁夏在0.3%以下。
【G6PD催化原理与功能】
G6PD是磷酸戊糖通路中的第一个催化酶,也是限速酶,单体由515个氨基酸残基组成,相对分子质量约59 000,二聚体和四聚体形式有催化活性。在人红细胞,主要以二聚体存在。G6PD的主要功能(特别在红细胞中)是生成潜在抗氧化剂还原型辅酶Ⅱ(NADPH)。NADPH为维持谷胱甘肽(GSH)还原状态所必需,还原型GSH可与过氧化氢(H2O2)和氧游离基反应,从而保持红细胞中血红蛋白和其他蛋白质如含巯基酶的还原状态(G6PD催化反应示意图见图5-2)。显而易见,G6PD缺乏将使红细胞不能维持还原状态,由此引发红细胞氧化性损伤而导致溶血。

图5-2 G6PD催化反应示意图
【遗传方式】
G6PD的基因定位于X染色体长臂α区δ带(xq28),遗传方式为X伴性遗传,不完全显性,具有不同的表现度。①男性患者为半合子,由于只有一条X染色体,故酶活力显著缺乏,此男性与正常女性婚配,所生儿子全部正常,女儿全部携带该缺陷基因,为女性杂合子。②女性有两条X染色体,女性杂合子的另一条X染色体等位基因正常,通常溶血代偿良好而无贫血,但是酶活力显著减低时也可表现临床症状。女性杂合子与正常男性婚配,有50%概率遗传给后代,获得突变基因的儿子有临床表现,女儿则有50%可能为杂合子。③由于G6PD缺陷发病率很高,所以女性纯合子并不罕见,可有严重的溶血表现。女性纯合子与正常男性婚配,理论上儿子100%为携带该缺陷基因的半合子,女儿100%为携带该基因的杂合子。
【G6PD缺乏症分型】
早先将G6PD缺乏症分为两类,即急性溶血类型和慢性溶血类型,随着对G6PD生化变异型和基因突变型的研究,WHO又将急性溶血型分为黑种人高发类型和地中海地区高发类型。近年根据基因型与临床表型关系的研究,以及酶活力与G6PD晶体三维结构突变位点的关联,将G6PD缺乏症分为5型,可以更好地理解其病理机制,这种分型称为变异型分型或多态性分型。另一种分型是以诱因和临床表现为主的临床表型分型,便于临床诊断,两种分析的对应关系见表5-2。
(一)G6PD缺乏症临床表型分型
分为5种类型:先天性非球形红细胞溶血性贫血(CNSHA)、蚕豆病、新生儿溶血、药物性溶血、感染性溶血。
(二)G6PD缺乏症变异型分型
Ⅰ型:即CNSHA,表现为慢性非球形溶血性贫血,溶血严重程度差异很大,从轻微溶血到依赖输血均见报道,以溶血严重者为多。Ⅰ型在G6PD缺乏症中较少见,散在发生。
Ⅱ型:表现为急性溶血,酶活力小于正常值的10%,症状严重。Ⅱ型即为WHO分型中的G6PD Mediterranean,多见于地中海人群如意大利、希腊、西班牙、阿拉伯及犹太(库尔德)后裔,亚洲包括我国等东南亚国家也是高发地区。此型可以引起新生儿溶血、药物性溶血和蚕豆病。
Ⅲ型:表现为急性溶血,酶活力为正常值的10%~60%,中等程度临床症状。Ⅲ型即WHO分型中的G6PD A-,非洲地区高发,非洲裔美国人中男性发病率为10%。该类型可以发生新生儿溶血、药物性溶血,但是很少因食用蚕豆而溶血。
Ⅳ型:临床症状轻度或无症状,酶活力为正常值的60%~150%,罕见。
Ⅴ型:有基因突变,无临床症状,酶活力增高,在正常值的150%以上,罕见。
【G6PD基因突变类型】
G6PD基因核酸长度2 269kb,含有13个外显子,12个内含子,编码区长度1 545kb,共18.5kb碱基对,编码515个氨基酸残基。1989年Kanno等人报道,人红细胞G6PD存在两种亚型,两者C端479个氨基酸残基相同,但是N端的排序和长短不同,编码主要亚基N端区域的基因位于6号染色体,即X染色体和6号染色体上两种不同的结构基因共同编码单链亚基。由两种mRMA交叉翻译而合成红细胞G6PD的机制尚不清楚,目前报道的突变基因均在X染色体上。
1966~1986年报道400种以上的G6PD生化变异型,G6PD有理化性质、酶代谢动力学、电泳行为等异常;1986~2006年鉴定出G6PD基因突变型在140种以上,一种基因突变可产生不同的生化变异型,这可能与该酶缺陷在不同人群中表现的多态性有关。
G6PD突变均发生在编码区即外显子,绝大多数是点突变,有5种小片段缺失,1种终止密码突变。G6PD B是野生型等位基因,一般正常人为B型。G6PD A+(电泳迁移率快于B)是正常黑人中显现率很高的非洲人种特异性多态等位基因,酶活性基本正常,无溶血。非洲人群G6PD A-(Val68Met、Asn128Asp)和地中海地区的G6PD Mediterranean(Ser188Phe)是发生率很高的G6PD缺陷变异型,还有东南亚一些高发变异型均可引起急性间歇性溶血。
地区性大样本分析各基因型发病率的研究结果表明,G6PD基因突变和频率差异很大,基因型有地域性突变热点。墨西哥14个州4 777人筛查G6PD缺陷,发病率为0.71%,76人缺陷鉴定出10种变异[A(202A/376G)、G6PD A(376G/968C)、G6PD Santamaria(376G/542T)、G6PD Vanua Lava(383C)、G6PD Tsukui(del561-563)、G6PD“Mexico City”(680A)、G6PD Seattle(844C)、G6PD Guadalajara(1159T)、G6PD Nashville(1178A)、G6PD Union(1360T)],其中G6PD Santamaria(376G/542T)变异体占82%,发病率1.64%。在西南太平洋Vanuatu archipelago地区测定1 442名男性,有98例(6.8%)G6PD缺陷,其中53例残余酶活力<10%的患者突变型相对集中在4种表型:G6PD Union[(C1360T(Arg 454Cys)]、G6PD Vanua Lava[T383C(Leu 128Pro)]、G6PD Namoru[T208C(Tyr70His)]、G6PD Naone[G497A(Arg 166His)]。非洲裔美国人中G6PD A+(A 376G)基因频率为11%,G6PD A(G 202A)基因频率为20%。
中国人G6PD基因突变类型与国外报道有显著区别。中国台北在1987年7月至1997年12月,从1 143个医疗单位采集足后跟血样2 971 192份,在4个新生儿筛查中心确诊46 570例G6PD缺陷,1997年新生儿筛查覆盖率99%,计算结果台湾发病率为2.1%(男性3.1%,女性0.9%)。台湾突变位点的基因频率分别为:46.8%(G1376T)、16.2%(G1388A)、7.9%(A95G)、6.5%(A493G)、5.6%(G392T)、4.6%(C1024T)、0.5%(G487A)、0.5%(C519G),与中国内地和中国香港地区及海外华裔报道的最常见突变型(G1376T、G1388A、A95G)和基因频率一致,证明中华民族统一起源。G1376T和G1388A是中国人特有G6PD突变型,此外,见于中国人的突变型还有C1387T、G1381A、G1360T、C1004T、G871A、A835T、A835G、C592T和T517C。
最常见的G6PD突变型及地理分布见表5-1。
表5-1 常见G6PD突变多态类型及地理分布

续 表

续 表

引自Mason PJ,et al.Blood Rev,2007,21(5):267。
【基因突变类型与临床表型的关联】
2000年前后获得人G6PD晶体并对一些突变进行三维结构分析,酶蛋白晶体可以展示蛋白质高级结构,更加有利于分析基因型与临床表型的关系。
从酶蛋白高级结构看,G6PD是由两个二聚体再聚合而成的四聚体构象。每个亚基都有底物G6P结合位点和辅酶NADP结合位点,一个亚基结合一分子NADP+,结合位点位于N端,第72位精氨酸(Arg72)与辅酶结合直接相关,是保守氨基酸,在23个生物种属保守性高度一致。一般而言,突变距离底物结合位点和辅酶结合位点越近,或突变点保守性越高,患者临床症状越严重。
G6PD高级结构研究表明,引起严重缺陷症的突变多发生在酶底物、NADP+结合位点附近和二聚体界面,这些催化性区域和结构性区域发生构象改变,影响G6PD功能。发生在疏水环内的突变也可影响酶蛋白二级结构。例如,Ⅰ型变异型即慢性非球形溶血性贫血突变多集中发生在G6PD基因的NADP结合位点附近。从基因序列上看,症状严重的Ⅰ型慢性溶血的突变几乎都发生在第10和第11外显子,即氨基酸序列380~430位,靠近二聚体界面,影响蛋白质折叠,影响疏水性和离子键结合力,酶蛋白表现为热稳定性明显下降。Ⅰ型虽然是散发突变,但431位为突变热点,如Sumare V431G和SurabayaV431M,两者临床表型有差异。急性间歇性溶血(Ⅱ、Ⅲ型)和无溶血(Ⅴ型)的变异型则散在发生于G6PD基因全长序列中,这些突变多影响NADP的结合和酶分子的稳定性,而稳定的二聚体是体内保持酶活性的关键。如G6PD A-型突变影响酶蛋白折叠,与野生型相比,在体外变性后复性时,重新折叠非常缓慢而且不完全,在体内酶的半衰期缩短。G6PD变异分型与基因突变的相关性参见图5-3。
G6PD缺乏一般只影响红细胞功能,如G6PD A-型红细胞残余酶活力仅有10%,白细胞酶活力却100%。但是有一些突变同时影响有核细胞,均为Ⅰ型CNSHA,红细胞残余酶活力小于1%时,白细胞酶活力可降至33%以下,例如在C514T(Pro172Ser)突变,可导致白细胞G6PD活力仅为正常值的3%,该荷兰患者G6PD严重缺乏、易反复感染、中性粒细胞爆发呼吸减弱,在体外培养的肌纤维母细胞中G6PD活力也明显下降,约为15%,Km(NADP/G6P)升高4~5倍。一例葡萄牙患者新变异C269Y突变的等位基因在面颊细胞中也存在,表明出现嵌和现象。这些突变型可能对G6PD缺陷与其他疾病相关性研究有提示意义。

图5-3 G6PD基因编码序列常见突变位点示意图
带数字空心矩形:外显子编号;空心圆形:Ⅱ型、Ⅲ型变异型的突变;实心圆形:严重溶血的Ⅰ型变异型散在突变;空心椭圆形:Ⅳ型突变;实心矩形:小片段缺失;X:无义突变;f:剪接位点突变;202A和968C:非洲人种常见的G6PDA-碱基置换位点;Mediterranean:地中海地区常见突变位点;Seattle、Union:常见突变,见于意大利南部、萨丁尼亚、希腊、阿尔及利亚、德国、爱尔兰、西班牙、葡萄牙、墨西哥等地区。Union亦见于中国
引自Fiorelli CG.Lancet,2008,371:64
【G6PD基因突变的进化优势】
由于G6PD缺陷高发区与疟疾高发区在地理上有引人注意的重合现象,国内外学者都对此进行了研究,一致的结论是G6PD缺陷红细胞对疟原虫入侵有抵抗力,这是突变基因在进化过程的优势选择。例如有G6PD A-等位基因的男性和女性恶性疟原虫感染的危险性分别下降46%和58%。我国疟疾高发区云南地区高频率的G6PD G1388突变可能与疟原虫抗性遗传优势有关,携此基因的无症状G6PD缺乏者具有抵抗疟疾能力。
G6PD缺陷红细胞抵抗疟原虫入侵的机制仍不清楚,已对缺陷红细胞膜的血型糖蛋白、离子交换蛋白、脂筏结构等进行了大量研究,形成多种推测机制。就红细胞本身代谢而言,缺乏G6PD的红细胞中微环境的改变不利于虫体生存,可能是其抗疟机制之一。
【G6PD缺乏症溶血机制】
G6PD缺乏所致溶血机制尚未完全明了。由于G6PD缺乏症在变异型上、在临床表型上有很大的差异,现有学说难以以一概全。尽管各型在溶血诱因、溶血场所、溶血过程有所不同,但发生溶血的根本原因相同,即还原型NADPH生成不足,使红细胞受到氧化损伤。
正常红细胞中G6PD只发挥其作用的2%,所以包括Ⅱ型和Ⅲ型变异型在内的多数患者平时可无临床表现,但是在氧化应激状态下,如服用氧化性药物时,正常红细胞磷酸戊糖通路代谢活性明显提高,至少在5倍以上,产生足量还原型NADPH以保护红细胞,而G6PD缺乏者由于酶活力低下不能快速生成足量NADPH,进而影响GSH生成,致使氧化性药物作用下所形成的H2O2等过氧化物不能被迅速还原而将GSH耗竭殆尽,过量的H2O2可将血红蛋白、酶蛋白、膜蛋白上的巯基(-SH)氧化交联为二硫键(-S-S-),膜磷脂也被氧化为过氧化磷脂,结果致使酶蛋白失活、血红蛋白变性为GSS-Hb和MHb,并与变性的膜脂和膜蛋白形成不可逆变性珠蛋白小体(Heinz bodies),膜变形性明显下降,易被单核-巨噬细胞系统吞噬破坏,或被脾脏摘除Heinz小体而形成缺失膜表面积的咬痕细胞(bite cells)。大量受氧化损伤的红细胞不能被脾脏等吞噬细胞及时处理,便发生血管内急性溶血。Ⅰ型先天性非球形红细胞溶血性贫血则以单核-巨噬细胞系统溶血为主,即表现血管外慢性溶血过程。
临床表型中感染性溶血的机制不明,可能感染时白细胞在吞噬过程释放氧化性物质,引起红细胞氧化应激反应而导致G6PD缺陷红细胞发生血管内急性溶血。蚕豆病的溶血机制较药物等诱因溶血更为复杂,蚕豆中的香豌豆嘧啶、异尿咪和伴蚕豆嘧啶核苷可能是致氧化性溶血的成分,通过不甚明确的氧化途径,使GSH减少而致溶血。
近年研究表明,G6PD缺陷还存在其他几种溶血机制,其中有些机制得到更多的实验支持。有实验显示,红细胞的氧化损伤并不依赖于GSH的浓度,而是与NADPH的浓度直接相关。NADPH与GSH过氧化物酶(GPx)催化反应(GSSG→GSH)和过氧化氢酶(触酶,CAT)催化反应(H2O2→H2O)均有密切关联,在清除过氧化物、保护红细胞的机制中,CAT起着比GPx更为重要的作用。对G6PD缺陷患者红细胞膜的研究显示,膜脂质中磷脂酰乙醇胺(PE)含量减少,膜脂质过氧化损伤的产物丙二醛(MDA)明显增高,膜蛋白受氧化的高分子聚合物增多。膜损伤不仅导致红细胞变形能力下降,而且诱发膜带3蛋白聚簇变化,形成衰老抗原,为自身抗体所识别,经由网状内皮系统吞噬,加速病变红细胞衰老死亡而产生溶血、贫血症状。
溶血自限性是G6PD缺乏症急性溶血的特点。G6PD是典型的细胞年龄依赖性酶,酶活力随着红细胞衰老而逐渐下降,衰老红细胞酶活力下降50%以上,G6PD缺陷的年轻红细胞不易受氧化损伤,但病态衰老红细胞对氧化物质很敏感,氧化损伤导致急性溶血发作时,衰老细胞可于数日内破坏殆尽,机体对红系代偿增生,产生大量年轻红细胞,溶血可自行减轻以至终止,这种现象称为溶血自限性。
【临床表现】
红细胞G6PD遗传性缺乏是产生一大类溶血的遗传背景,在G6PD缺乏的基础上,不同诱因可以产生临床表现和溶血机制有差异的溶血类型,临床分为5型,即先天性非球形溶血性贫血、新生儿黄疸、蚕豆病、药物性溶血和感染性溶血。后4型还存在尚无定论的其他病因,与单纯G6PD缺乏有区别。以溶血诱因区分的5种临床表型和以酶活力区分的五种多态性变异型在临床表现上可以部分重合(表5-2),为便于诊断治疗,多根据溶血诱因分述这些病症。
表5-2 G6PD缺乏症各型临床表现

1.先天性非球形红细胞溶血性贫血(congenital nonspherocytic hemolytic anemia,CNSHA) 即以慢性溶血为表现的G6PD缺乏症,多态性变异型为I型,较少见。溶血场所和溶血方式与其他类型明显不同,CNSHA的特点是血管外慢性溶血为主,有脾脏肿大、网织红细胞增高、胆红素和乳酸脱氢酶升高、胆结石。慢性溶血状态受到氧化应激可加重溶血,常要输血。许多患者有严重的新生儿溶血史,但在婴幼儿期一直疑诊,可能与急性溶血期红系代偿增生导致检测指标假阴性有关。
2.新生儿黄疸 通常在出生1~4天发现黄疸,与生理性黄疸类似,但是持续时间比血型不相合导致的黄疸期明显延长,黄疸过高者需予以治疗以防发生核黄疸而造成神经系统永久损伤。G6PD缺陷的早产儿比足月出生的新生儿黄疸更明显、溶血更加严重。如果患儿同时合并可导致Gilbert综合征的遗传缺陷(尿嘧啶核苷二磷酸葡萄糖苷酸转移酶(UGT1A1)基因启动子突变),将发生严重的新生儿黄疸,甚至危及生命。临床判断:出生后总胆红素高于150μmol/L,或者同胞中有新生儿黄疸史。
3.蚕豆病 通常食用蚕豆后24h溶血发作。尽管胆红素水平不一定会很高,但是溶血程度重于药物和感染诱因溶血。患者有明显的血管内急性溶血症状,表现为贫血、黄疸及典型的酱油色血红蛋白尿又称茶色尿。重者可出现溶血危象,表现为急速贫血、畏寒、发热、恶心、呕吐、口渴、腹痛、腰痛、背痛等。极重型患者病情发展迅速,可出现神志不清、抽搐甚至休克。血红蛋白管形尿或肾脏局部缺血造成肾脏损伤,也可以导致急性肾功能衰竭。如果对症处理不及时,会危及生命。蚕豆病性溶血导致的氧化损伤可促发循环系统清除机制,因此患者既有血管内溶血,又有血管外溶血,溶血期间脾脏可有明显肿大。临床病例表明,并非所有G6PD缺陷者食用蚕豆后都会发生溶血,在同一个体也不是每次食用蚕豆都会溶血发作。溶血发作与否和溶血严重程度除了取决于G6PD残余活力外,还取决于患者身体状况、蚕豆食用量以及其他多种因素。新鲜蚕豆比干燥蚕豆和冷冻蚕豆更易导致溶血,可能鲜蚕豆中氧化性的香豌豆嘧啶、异尿咪和伴蚕豆嘧啶核苷含量更高。母亲食蚕豆后哺乳患儿,也可导致患儿溶血。笔者诊断1例食用蚕豆无碍但食用菠萝诱发溶血的G6PD缺乏症,提示食物中并非仅蚕豆为溶血诱因。已报道的其他可能引起急性溶血的食物还有红酒、开胃水(tonic water,含奎宁汽水)、蓝梅或含有蓝梅的酸奶制品。
4.药物性溶血 一般在服药24~72h溶血发作,血红蛋白尿所致酱油尿或茶色尿是一明显特征。贫血症状加重直至到第7~8天,停药后8~10天Hb开始恢复。该类溶血Heinz小体生成试验阳性。影响药物性溶血严重程度的因素很多,先天性因素中包括细胞的代谢状态、G6PD酶缺陷类型、药物代谢动力学的遗传差异等,后天性因素中包括年龄、性别、药物的剂量、吸收、代谢和排泄,药物及代谢产物对酶活力的影响、感染存在对氧化应激的强化作用、服药时血红蛋白原有浓度以及红细胞年龄群的分布等。导致G6PD缺乏症溶血的常见药物见表5-3,这些药物分子中多含有氧化性基团。如有感染可增加酶缺陷细胞对氧化性药物的敏感性。
5.感染性溶血 感染是最典型的溶血诱因,可引起红细胞氧化应激反应,具体机制不明,可能与感染时白细胞吞噬功能加强、释放氧化性物质增多有关。常见感染包括肺炎、流感、伤寒、甲肝乙肝病毒、巨细胞病毒、沙门菌、大肠埃希菌、β-溶血性链球菌、立克次体等感染。溶血、黄疸的严重程度与当时使用药物的种类、肝脏功能、年龄等多种因素有关。对重症溶血者立即输血可以快速改善临床进程。合并病毒性肝炎溶血患者的潜在并发症是致命的急性肾衰竭,这是由于血红蛋白尿形成的管形结晶使肾小管梗阻、缺血而导致急性肾小管坏死,有些患者需要透析治疗。感染性溶血急性肾衰竭在幼儿很少见。
【诊断】
G6PD缺乏症以急性溶血表现为多见,任何年龄均可发病,男性症状明显,临床诊断时应特别注意有无溶血诱因和相关病史及家族史、有无酱油尿。实验室确诊指标是直接测定红细胞G6PD酶活力。
(一)临床诊断
1.有溶血诱因
(1)蚕豆:近期(通常1~2天内,最长可在1周左右)食用蚕豆或蚕豆制品;母亲食用蚕豆后,患儿吸吮其母乳而发病;蚕豆开花期接触或吸入花粉也是潜在诱因。
(2)感染:近期患病毒或细菌性感染,如病毒性呼吸道炎、肺炎、传染性肝炎、传染性单核细胞增多症、伤寒、败血症等。
(3)药物:近期使用药物后出现急性溶血症状。小儿接触含萘防虫剂(樟脑丸等)保存的衣物、被褥后发生溶血。
(4)其他诱因:过强体能锻炼、过度疲劳、糖尿病、心肌梗死等也可促发患者急性溶血。
2.有相关病史、家族史 患者或其血缘亲属有新生儿黄疸史、有CNSHA和(或)急性溶血史。患者籍贯多见于华南、西南地区。
表5-3 G6PD缺乏症禁用、慎用的药物和避免接触的化学试剂

*:对地中海、亚洲变异型高危;**:溶血高危、低危报道不一致。
3.有溶血症状
(1)慢性溶血症状:CNSHA有血管外慢性溶血表现,不同程度的贫血、脾肿大、网织红细胞增高、未结合胆红素增高,可有胆结石等并发症。在有感染、用药等溶血诱因存在时,可以加重溶血症状。其他类型的G6PD缺乏症无溶血诱因时一般无溶血指征,少数蚕豆病患者平时可有轻度胆红素升高,脾肿大不明显。女性纯合子可有CNSHA表现。
(2)急性溶血症状:最典型的症状是短期内出现贫血、疲倦、酱油尿。上述各种诱因导致G6PD缺乏者发生急性血管内溶血,发病的潜伏期越短,症状越严重,主要表现为血红蛋白和红细胞计数急剧下降,出现皮肤巩膜黄染,尿液呈酱油色或浓茶色,可伴有畏寒、发热、呕吐、腰腹疼痛等。严重溶血者特别是并存肝肾疾患者可出现溶血危象或肾衰竭。蚕豆病患者在急性溶血发作期间脾脏可能出现轻度增大。
(3)性别差异:发生急性溶血的患者以男性多见,家系中男性患者症状明显严重。女性纯合子可表现为CNSHA,女性杂合子中约有10%的人可发生急性溶血。
4.有溶血自限性 G6PD缺乏症所致急性溶血的特点是具有自限性,即当溶血达到一定程度,引起溶血的诱因虽未解除,溶血过程不再发展,恢复过程长短与患者酶缺乏程度有关。
(二)实验诊断
G6PD酶活力定量测定是特异性的确诊指标,定性分析和非特异性指标可以辅助诊断。
1.符合溶血指征 尤其是血红蛋白尿检阳性,是G6PD缺乏症急性发作时血管内溶血的诊断依据。
2.红细胞形态学 急性溶血期外周血红细胞形态可有一些非特异性的改变,见红细胞大小不等、有核红细胞、嗜多染性红细胞和红细胞碎片,也可见少量口形、棘形红细胞。部分患者红细胞涂片可见少量“咬痕”细胞(图5-4)。

图5-4 G6PD缺陷患者“咬痕”细胞
3.特异性诊断指标
(1)G6PD活力定量测定:这是确诊依据,直接定量测定红细胞G6PD催化反应的产物NADPH含量。男性患者酶活力大多显著下降,国内男性患者酶活力多在正常值的10%以下,应归为G6PDⅡ型变异型。女性杂合子酶活力下降不明显,女性纯合子酶活力可表现明显低下。
G6PD是年龄依赖性酶,细胞越年轻,酶活力越高,网织红细胞的G6PD活力比衰老红细胞要高5倍,所以含大量年轻红细胞的新生儿可能酶活力测值偏高而部分掩盖G6PD缺陷。在溶血发作期、恢复初期、近期输血等状态下检测酶活力,可因患者高网织红细胞或外源正常细胞影响,不能反映出真实酶活性。解决方法:做G6PD/6PGD比值纠正、低渗溶血测值纠正,或3个月后复查酶活力。
(2)G6PD活性定性分析
1)荧光斑点试验:作为初筛试验。NADPH在长波紫外线照射下能显示荧光,G6PD缺陷的红细胞内NADPH生成量少,荧光减弱,以此推测G6PD活性。G6PD严重缺乏者30min不出现荧光,中度缺乏者在10~30min之间出现荧光,而G6PD活性正常者10min内即出现荧光。高网红、溶血发作期、新生儿、女性等因素可导致假阴性。
2)硝基四氮唑蓝纸片法:反应滤纸片在G6PD活性正常者、中度缺乏者和严重缺乏者分别呈紫蓝色、淡紫蓝色和红色。
3)红细胞G6PD洗脱染色法:作为筛查试验。缺乏G6PD的红细胞中高铁血红蛋白不能立即被还原,遇氰化物后形成氰化高铁血红蛋白,易被过氧化氢洗脱,致使红细胞经染色后呈不着色的空影。正常人空影红细胞<10%,G6PD杂合子约50%,纯合子在75%以上。适用于女性杂合子的检出。
(3)酶变异型分析(1967年WHO推荐分析指标):酶活力、电泳迁移率、对G6PD和NADPH的Km、同类底物利用率、酶的耐热性和最适pH。
(4)基因突变型分析:使用限制性酶切、PCR、变性梯度胶电泳、直接测序等方法,可以鉴定G6PD基因突变类型和多态性,也有进行产前诊断的研究。由于尚不能在临床常规应用,所以不能列为G6PD缺乏症的诊断标准。
4.非特异性诊断指标
(1)高铁血红蛋白还原试验:以高铁血红蛋白还原率间接反映磷酸戊糖旁路代谢状态,作为G6PD缺陷初筛试验。G6PD显著缺陷者<30%,中间型为31%~74%,正常人>75%。在血红蛋白H、不稳定血红蛋白病、高脂血症及巨球蛋白血症等可出现假阳性。
(2)变性珠蛋白(Heinz)小体生成实验:G6PD缺陷的红细胞内NADPH生成减少,导致GSH减少,内环境不稳定,在氧化剂盐酸苯肼作用下,血红蛋白等含巯基物易受氧化,变性沉着于膜上,形成Heinz小体。G6PD显著缺陷者阳性细胞>28%,正常人<28%。
Heinz小体成因不限于G6PD缺陷,不稳定血红蛋白的α链或β链中发生氨基酸置换或缺失,使分子不稳定,也易受氧化形成Heinz小体。珠蛋白生成障碍性贫血红细胞中血红蛋白肽链综合不平衡,过剩的α链或β链沉积于膜上也可形成Heinz小体。因此本实验无特异性。有学者将Heinz小体阳性的病统称为Heinz小体病,包括G6PD缺陷、GR缺陷、GSH缺乏、不稳定血红蛋白病、珠蛋白生成障碍性贫血、血红蛋白lepore等。
(3)GSH含量测定:G6PD缺乏患者通常测值为正常值的60%~78%,蚕豆病现症者在50%以下。GPI缺乏及不稳定血红蛋白变异体都有GSH中度下降,涉及GSH代谢的酶有缺陷时,GSH含量亦下降。
【鉴别诊断】
(1)G6PD缺乏症中Ⅰ型CNSHA临床症状同糖酵解等代谢酶缺乏,均为慢性溶血,有脾大、胆结石等症状,直接酶活力测定予以鉴别。
(2)有些G6PD缺陷症患者合并另一种可导致溶血或黄疸的红细胞遗传缺陷,症状严重,非急性溶血发作期可持续有高网织红细胞、高胆红素、脾大等慢性溶血表现,须做溶血系统分析和家系调查予以排查和确诊。已报道的合并疾病有遗传性球形红细胞增多症、珠蛋白生成障碍性贫血、GPI缺陷、PK缺陷、先天性红细胞生成不良性贫血(CDA)和Gilbert综合征等。
(3)发生急性血管内溶血的遗传因素还包括红细胞氧化还原代谢酶缺陷,需做特异性酶活力鉴定。
【治疗方案及原则】
无特效对因疗法。以饮食与药物预防为主,禁食蚕豆及其衍生制品,注意防感冒、防感染、慎用药。一般处理原则见“红细胞酶病概述”章节。
(一)特殊处理:溶血期支持疗法
1.溶血危象期 输血、输液,抗生素控制感染,防治休克与急性肾衰竭。注意确保输注血源无G6PD缺陷,否则会导致第二次溶血。
2.新生儿黄疸的治疗 要控制高胆红素血症,以预防核黄疸的发生。总胆红素>150μmol/L需做光照疗法,>300μmol/L须输血换血治疗。注意避免使用氧化性药物,纠正缺氧、酸中毒和防治感染。
(1)光照疗法:分解未结合胆红素,促进转化产物排出体外。用波长420~440nm蓝色荧光灯光照,转变体位,连续24~72h,直至黄疸明显减退(<140μmol/L)。光疗的不良反应有腹泻、脱水及青铜症等,应注意补液。光疗期间应补充核黄素,因维生素B2易受光氧化破坏,致使GSH还原酶活性降低,可加重溶血。
(2)换血疗法:起效快,适用于核黄疸早期、血清总胆红素超过≥300μmol/L者(国内标准多为>200μmol/L)。
(3)药物治疗:促胆红素转化、结合与排泄。常选用药物苯巴比妥、白蛋白、10%葡萄糖等制剂和中药复方茵陈蒿汤。
(二)产前预防性用药
G6PD缺乏的孕妇,于产前2~4周在医生指导下小剂量服用鲁米那(苯巴比妥),诱导肝内产生胆红素代谢相关酶,对减轻新生患儿出生后的高胆红素血症、预防核黄疸具有一定作用。
(三)脾脏切除术
G6PD缺乏症绝大多数为Ⅱ型、Ⅲ型多态性变异型,以血管内急性溶血为主要表现,脾脏无明显肿大,因此脾脏切除对大多数病例无效。脾切除对CNSHA慢性溶血脾大者的改善作用不明显,对脾肿大明显的女性纯合子有改善作用,可减少输血或无需输血。
(四)抗氧化剂
维生素E和硒制剂对CNSHA慢性溶血者有改善作用,对其他类型无明显作用。
(五)G6PD缺乏症用药禁忌
常见的可诱发G6PD缺陷者发生溶血的药物见表5-3。
二、谷胱甘肽代谢相关酶
谷胱甘肽(GSH)的代谢涉及多种反应途径多种代谢酶,与遗传性溶血性贫血有关的缺陷酶主要有4种,即谷胱甘肽还原酶(GR)、谷胱甘肽过氧化物酶(GPx)、γ谷氨酰半胱氨酸合成酶(GCS)和谷胱甘肽合成酶(GSS)。GR和GPx参与GSH氧化还原反应,GCS和GSS参与GSH的合成反应。γ谷氨酰循环是GSH合成与分解反应及细胞外氨基酸转运至细胞内的代谢途径(图5-5),某些中间代谢产物与酶缺陷病理表现有关。
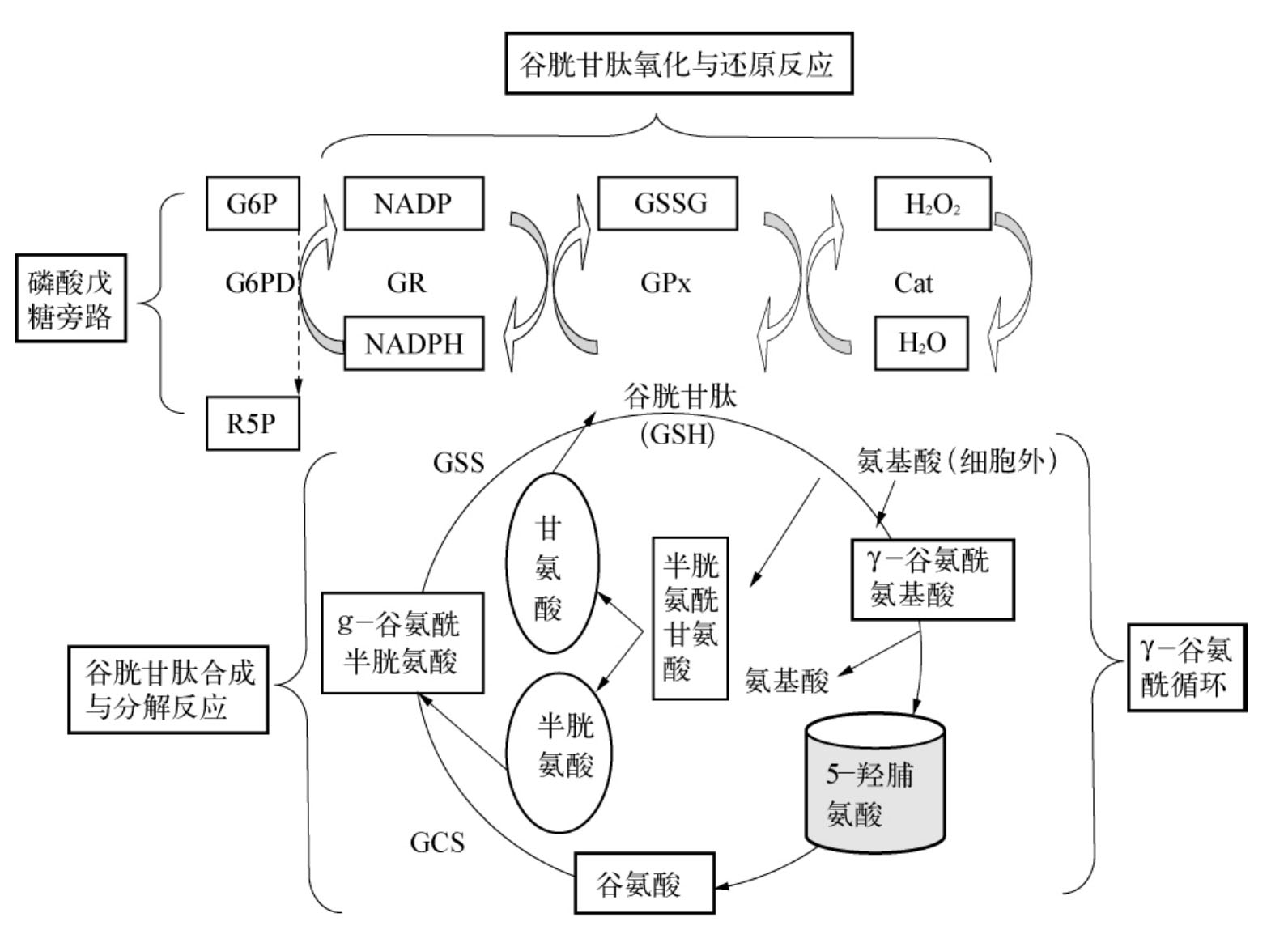
图5-5 谷胱甘肽代谢相关反应及γ-谷氨酰循环
GSH是体内重要的抗氧化剂,可保护机体免受过氧化物的损伤,对维持含巯基的酶和蛋白的活性极为重要。GSH对红细胞更为重要,因为成熟红细胞无细胞核和细胞器,不能再进行蛋白合成反应,所以要维持原有酶和蛋白特别是血红蛋白、膜蛋白的功能状态,依赖于GSH的抗氧化保护作用。无论哪一种GSH代谢酶缺陷,结果都会影响GSH的含量和功能,引起溶血性贫血,有些酶缺陷还可累及其他系统。
上述酶缺乏症的共性是符合CNSHA诊断,GSH水平低下,Heinz小体生成试验可以呈阳性,氧化性药物可诱发溶血。确诊指标必须是各个酶的活力测定和(或)基因突变位点鉴定。
(一)谷胱甘肽还原酶(glutathione reductase,GR,EC 1.6.4.2)
GR使氧化型谷胱甘肽(GSSG)还原为还原型谷胱甘肽(GSH)。GR缺陷分为遗传性与继发性两种。遗传性GR缺陷为常染色体显性遗传,临床症状为慢性溶血,可伴有全血细胞减少、智能发育不全、肌强直、白内障等。继发性缺陷又称核黄素缺乏型,GR含量可降至正常值的38%~60%,在氧化性药物等诱因作用下发生溶血,口服核黄素后GR可以恢复正常。
(二)谷胱甘肽过氧化物酶(glutathione peroxidase,GPx,EC 1.11.1.9)
GPx参与GSH向细胞内过氧化物质递氢反应,解除过氧化物对细胞的损伤,维持细胞内氧化还原平衡。GPx遗传方式为常染色体隐性遗传,酶缺陷杂合子出生时可发生严重的新生儿溶血。有些患者成年后无明显临床症状,但药物(如抗癫 药卡马西平)可诱发血管内急性溶血,患者有嗜碱性点彩增多,Heinz小体试验阳性。
药卡马西平)可诱发血管内急性溶血,患者有嗜碱性点彩增多,Heinz小体试验阳性。
(三)γ-谷氨酰半胱氨酸合成酶(γglutamyl-cysteine synthetase,GCS,EC 6.3.2.2)
GCS催化谷氨酸与半胱氨酸合成谷氨酰半胱氨酸,是γ谷氨酰循环限速酶。GCS缺乏症遗传方式为常染色体隐性遗传,表现为轻至中度溶血性贫血,成年时出现进行性脊髓小脑变性症状,男性还有肌无力、腱反射消失、语言断续等症状。在一例近亲婚配家系,先证者完全缺乏GSH,已鉴定其GCS突变基因在外显子11上1241C>T(Pro414Leu)。
(四)谷胱甘肽合成酶(glutathion synthase,GSS,EC 6.3.2.3)
GSS催化谷氨酰半胱氨酸与甘氨酸合成为GSH,在γ谷氨酰循环中GSS是最常见的缺陷酶。GSS缺乏症为常染色体隐性遗传,临床有两种表现:一种仅有溶血性贫血,临床表现差异很大,有些患者溶血代偿良好,贫血轻度,虽然网织红细胞明显升高,但肝脾肿大不明显,患者Heinz小体生成增多,药物和蚕豆可诱发急性溶血。另一种除溶血外,GSS缺陷累及多系统,大量5-羟脯氨酸出现于血和脑脊液中,产生5-羟脯氨酸尿、严重代谢性酸中毒、中枢神经系统功能障碍、易受细菌感染,约25%在儿童期死亡。在γ谷氨酰循环中,由于GSS缺乏而使GSH合成减少,γ谷氨酰半胱氨酸合成增加,进一步反应受阻,使上游中间产物5-羟脯氨酸(又名焦谷氨酸)蓄积,当生成速率超过分解能力时,导致5-羟脯氨酸尿和代谢性酸中毒。
GSS缺乏症已经鉴定出30余种突变型,有错义、插入、缺失等突变,如T808C、A656G。所有移码突变、终止突变和异常剪接突变均可导致溶血伴发5-羟脯氨酸尿和神经发育损伤。治疗上维生素C和维生素E有助于提高患者体内抗氧化物水平,碳酸氢钠用于纠正酸中毒。
三、细胞色素b5还原酶
细胞色素b5还原酶(cytochrome b5reductase,b5R,EC 1.6.2.2)旧称高铁血红蛋白还原酶(NADH-methemoglobin reductase,MR),或黄递酶(NADH-diaphorase)。与其他遗传性红细胞酶病不同,b5R遗传缺乏纯合子临床症状不是溶血性贫血,而是高铁血红蛋白血症,表现为发绀。
【酶催化机制及发病机制】
b5R是黄素酶(脱氢酶-电子转移酶)家族成员之一,以胞质形式和膜绑形式参与多种代谢反应。b5R基因位点(DIA1)在22号染色体(22q13-qter),编码基因31kb,9个外显子,8个内含子,单一基因可产生多种转录本,即由不同启动子和不同的翻译起始编码同一基因,产生同一酶蛋白的多种同工酶形式,酶蛋白相对分子质量约为32 000。红细胞中既有胞质同工酶,也有膜绑同工酶,胞质酶与血红蛋白还原有关。成年人红细胞膜绑酶占b5R的20%~25%,婴儿胞质酶很低,所以膜绑酶对婴儿非常重要。
机体在生理条件下进行脱氧反应时,一些氧分子脱离血红蛋白成为超氧游离基(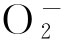 ),可将血红素结合的铁从二价形式(Fe2+)氧化为三价形式(Fe3+),即产生高铁血红蛋白(methemoglobin,MetHb),这是血红蛋白自动氧化作用,每天大约3%血红蛋白发生自动氧化。血红蛋白中的铁氧化状态决定其携氧能力,去氧血红蛋白二价铁形式易结合氧,反之MetHb中三价铁形成的高铁血红素不能与氧结合。内源性酶还原机制使MetHb低于血红蛋白总量的1%,MetHb生成增多或还原减少均可使MetHb水平升高而产生高铁血红蛋白血症(hemoglobinemia)。
),可将血红素结合的铁从二价形式(Fe2+)氧化为三价形式(Fe3+),即产生高铁血红蛋白(methemoglobin,MetHb),这是血红蛋白自动氧化作用,每天大约3%血红蛋白发生自动氧化。血红蛋白中的铁氧化状态决定其携氧能力,去氧血红蛋白二价铁形式易结合氧,反之MetHb中三价铁形成的高铁血红素不能与氧结合。内源性酶还原机制使MetHb低于血红蛋白总量的1%,MetHb生成增多或还原减少均可使MetHb水平升高而产生高铁血红蛋白血症(hemoglobinemia)。
高铁血红蛋白还原为血红蛋白有数种可能的机制(表5-4),而生理意义最重要的是NADH依赖性的细胞色素b5还原酶(b5R)催化反应。该酶分子中含有非共价结合的黄素腺嘌呤二核苷酸(FAD)作为电子递体,由糖酵解和糖醛酸代谢产生的NADH将FAD还原为FADH2,还原型的FADH2再将细胞色素b5还原,经由还原型细胞色素b5将电子传递给MetHb,使高铁还原为亚铁状态。在红细胞中,b5R将电子直接传递给MetHb使其还原。在有核细胞和网织红细胞中,b5R将电子传递给内质网硬脂酰辅酶A去饱和酶,参与胆固醇合成、脂肪酸延长与去饱和以及药物代谢等反应(图5-6)。
表5-4 红细胞中MetHb还原系统


图5-6 细胞色素b5还原酶催化反应
MetHb还原的旁路途径可以通过NADPH黄递酶催化,该酶的底物是NADPH,由G6PD催化戊糖旁路产生。NADPH作为电子供体可以在糖醛酸代谢中将氧化性的NAD+还原而生成NADH。NADPH还可以还原具有氧化还原性质的染料如亚甲蓝、黄素,还原型的亚甲蓝即无色亚甲蓝再将MetHb还原。因为这些电子受体不是生理性的,所以正常条件下电子转移作用不明显,NADPH黄素还原酶缺乏也不会引起高铁血红蛋白血症。但是,在急性中毒性高铁血红蛋白血症抑制了Cb5R的作用,亚甲蓝可起到治疗作用。需注意G6PD缺乏症者NADPH生成受阻,所以当患急性中毒性高铁血红蛋白症时,亚甲蓝治疗无效,此外由于亚甲蓝可以引起溶血,存在潜在危险。
MetHb也可以由抗坏血酸、还原型谷胱甘肽和还原型黄素直接还原,但是这些反应发生缓慢且作用有限。
目前已报道500余例b5R缺陷导致的高铁血红蛋白血症,已经鉴定出30余种b5R基因突变型,其中5例中国人变异型不同于国外报道。患者中近亲婚配家系占一定比例,其中一例先证者还有假两性体,该患者b5R基因发生剪接突变,终止密码提前,酶蛋白被截短。基因型b5R(116T>S)不会引起发绀,是非洲人种高频率的多态现象。b5R缺乏症突变型和临床表型大致可分为两类:Ⅰ型b5R缺陷通常为错义突变,以正常速率产生性质不稳定的异常b5R基因,有核细胞可以通过合成酶蛋白进行代偿,而红细胞酶活力明显下降。Ⅱ型b5R突变影响到催化位点或剪接异常、缺失突变导致酶结构改变,使酶蛋白合成量减少而减低酶活力,所有细胞酶活力都减弱。
【临床表现】
b5R缺乏症为常染色体隐性遗传,主要有两种临床表型:Ⅰ型b5R缺乏症仅红细胞酶活力下降,表现为发绀;Ⅱ型b5R缺乏症所有细胞b5R活力均下降,不仅有发绀表现,还有神经系统损伤。另有两例患者被定义为Ⅲ型b5R缺乏症,因为他们不仅红细胞缺乏酶活性,白细胞和血小板中酶活力也低下,但是没有神经系统症状。
Ⅰ型b5R缺陷在世界各地均有散在报道,但在某些人群中发生率较高,如北美阿撒巴斯卡印第安人、北美西部那瓦交印第安人和西伯利亚雅库次克本土人,是否为地方病仍有争议。纯合子或复合型双杂合子的高铁血红蛋白浓度升高至血红蛋白总量的10%~40%,出现发绀,但通常无症状,不影响寿命,孕期正常。有时观察到血红蛋白浓度明显代偿性增高,呈红细胞增多症。杂合子b5R活力约为正常的50%,平时无发绀,在正常状态下足以维持高铁血红蛋白在正常水平,但是对氧化性药物比正常人敏感,在氧化应激状态下超出其红细胞还原MetHb能力时,可产生急性高铁血红蛋白血症,出现发绀。
Ⅱ型b5R缺陷约占b5R缺乏症的15%,世界各地均有偶发,主要症状除了发绀以外,还出现严重的神经系统异常和发育延迟,包括智力障碍、小头畸形、角弓反张、手足徐动症样运动、斜视、癫 发作及痉挛性四肢轻瘫。由于神经系统合并症使预期寿命明显缩短,典型病例多在婴幼儿期死亡。病理机制尚不明确,可能涉及中枢神经系统脂肪延伸与去饱和异常。
发作及痉挛性四肢轻瘫。由于神经系统合并症使预期寿命明显缩短,典型病例多在婴幼儿期死亡。病理机制尚不明确,可能涉及中枢神经系统脂肪延伸与去饱和异常。
【诊断与鉴别诊断】
(一)b5R缺乏症诊断
1.临床诊断 发绀为b5R缺乏症纯合子首诊表现,患者自幼表现发绀,个别直到青春期才发生发绀。血浆MetHb含量超过15g/L就可表现发绀,唇、舌和上肢末端青紫色最为明显。患者平时活动正常,在运动加剧时可出现疲劳、气喘,氧化性药物、毒物可加重症状。患者父母及家系中b5R缺乏症杂合子平时无发绀,氧化性药物等氧化应激状态可诱使MetHb升高而致发绀。Ⅱ型b5R缺乏症除发绀外,还伴有神经系统病变,常见发育延迟、智力障碍、手足徐动症样运动、肌张力增高,少数患者有小头畸形、斜视、癫 ,个别报道有假两性体。低龄患儿可因病情加重而死亡。
,个别报道有假两性体。低龄患儿可因病情加重而死亡。
2.实验诊断
(1)血气分析:动脉血氧分压(PaO2)正常。此项目可与心肺功能异常导致的高铁血红蛋白血症鉴别。
(2)血象:血红蛋白含量(Hb g/L)可因机体缺氧而代偿性增高,MetHb超过血红蛋白总量的20%~50%的患者有轻度红细胞增多症。新鲜血液呈现深褐色。
(3)血浆高铁血红蛋白(MetHb)浓度:正常值<15g/L。发绀患者MetHb明显增高。
(4)血红蛋白光谱分析:患者未加酸处理溶血标本最大吸收峰光谱波长(λmax)与正常人相同,在415nm。加酸处理标本区别在940nm处,MetHb吸收峰强于正常血红蛋白和其他异常血红蛋白。631nm也有一个吸收峰。
(5)红细胞b5R活力测定:定量确诊指标,纯合子酶活力明显下降,杂合子酶活力约为正常值的50%。
(6)其他试验:①其他血细胞(白细胞、血小板)和体细胞(羊水成纤维细胞)b5R活力测定,有助于诊断Ⅱ型b5R缺乏症。②b5R的基因突变鉴定。
(二)引起高铁血红蛋白血症和发绀的其他原因
常见先天性原因有先天性心肺疾病、遗传性血红蛋白M病,常见的后天原因有药物性和毒物性高铁血红蛋白血症、心肺功能衰竭,还有年龄因素所至婴幼儿高敏体质。
1.先天性或后天性的循环系统或呼吸系统疾病所致高铁血红蛋白血症 这类疾病均可导致动脉血氧分压降低,还伴有其他心肺症状,容易与b5R缺乏症鉴别。
2.遗传性血红蛋白M病(Hb M病) 遗传方式与b5R缺乏症不同,Hb M病为常染色体显性遗传,患者多为杂合子,有明确家族史,运动耐量接近正常,不影响寿命,一般无需治疗。在导致MetHb增高的遗传因素中,b5R缺乏症发病率高于Hb M病。Hb M病珠蛋白链变异,使血红蛋白结构改变,所结合的铁不能从三价还原为二价,并且抑制Hb与氧结合,遂产生高铁血红蛋白血症与发绀。β链突变者出生数月后出现发绀,可伴有轻度溶血表现,α链突变者出生时即有发绀但无溶血。Hb M病诊断:①Hb电泳:淀粉凝胶电泳(pH 9)可见异常Hb区带。其他因素所致发绀血红蛋白电泳正常。②Hb吸收光谱测定:扫描波峰,除了有MHb吸收光谱特征外,不同突变的Hb可有不同于正常Hb的特殊吸收峰。
3.低幼儿血红蛋白血症 除了遗传性的b5R缺乏症和Hb M病以外,正常新生儿、婴儿也可能在接触氧化性药物、毒物时出现发绀,婴儿的C5bR活力仅为正常成人的50%~60%,早产儿活力更低,对氧化应激更加敏感。接触毒物的腹泻婴儿也会患高铁血红蛋白血症,机制不明,可能是内源性亚硝酸盐生成增加。可做家系调查、血红蛋白病检查或在少儿期后复查C5bR活力,排除遗传因素。
4.复合型双杂合子b5R缺乏症 如果患者双亲一方是b5R缺陷的杂合子;另一方是另一种红细胞遗传缺陷杂合子(红细胞膜病或其他类型红细胞酶病或血红蛋白病),均遗传给患者,则该患者为复合型双杂合子,其b5R活力为杂合型表现,可以不出现发绀而主要表现溶血症状,也可以表现发绀而溶血症状轻微。对此必须进行家系分析才能对病因确诊。
5.药物性或毒物性高铁血红蛋白血症 在获得性高铁血红蛋白血症中,以药物中毒所致多见,是剂量相关性还是特异体质所致尚无定论。药物、化工原料、化妆品成分、腐烂蔬菜等都含有致病成分,已报道的药物和化学品主要见于解热镇痛药(对乙酰氨基酚、乙酰苯胺、非那西丁)、麻醉药(苯佐卡因、利多卡因、丙胺卡因、氧化亚氮)、抗疟药(伯氨喹)、激素类(氟他胺)、抗微生物药物/抗麻风病药(磺胺甲猜唑、氨苯砜)、抗肿瘤药物(异环磷酰胺)、消化系统药物(甲氧氯普胺)、循环系统药物(硝酸甘油、亚硝酸异戊酯)、泌尿道消炎镇痛药(非那吡啶)、苯胺染料、亚硝酸盐(亚硝酸钠、亚硝酸异丁基)、硝酸盐(蔬菜多含,肠道细菌可将其转化为亚硝酸盐)、硝基苯、硝基乙烷(指甲膏清洗剂)、硝基呋喃类药、双吡啶化合物(对草快/绿谷隆,除草剂成分)等。这些药物、试剂也可引起溶血性贫血。
(三)鉴别诊断要点
1.发病 与b5R缺乏症相比,药物性和中毒性发绀起病急,症状重,组织急性缺氧,危及生命。症状的严重程度与血红蛋白向组织释氧能力损伤程度有关。早期症状包括发绀、头痛、疲倦、嗜睡、恶心、呕吐、心律增快及呼吸困难,少数可有发热。在MetHb较高水平,出现呼吸抑制、意识模糊、心力衰竭、休克、癫 样抽搐甚至死亡。
样抽搐甚至死亡。
2.病史 有药物或毒物接触史,中毒性MetHb血症可突发或集体发病。
3.动脉血氧分压 b5R缺乏症该指标正常,与心肺疾病发绀有别。
4.Hb光谱 硫化物中毒时产生硫高铁血红蛋白,浓度超过5g/L即出现发绀,血液呈蓝褐色,新鲜血样硫高铁血红蛋白特征吸收峰波长在620nm和654nm。在618~631nm处测定,标本中加氰化钾后,硫血红蛋白血620nm吸收峰不变,MetHb 631nm吸收峰迅速消失。
5.治疗反应 应用亚甲蓝及维生素C后,药物性所致MetHb血症发绀快速消退,对b5R缺乏症可改善发绀,对血红蛋白M病及硫血红蛋白血症均无效。硫血红蛋白血症的发绀表现在去除病因3个月后自动消失。
【治疗】
遗传因素引起的高铁血红蛋白血症如果不影响生命活动一般无需治疗,发绀严重、气急者可药物治疗。
1.b5R缺乏症 ①禁用可引起高铁血红蛋白血症的药物。②治疗药物:口服亚甲蓝,100~300mg/日,或口服维生素C,100~300mg,3次/日。维生素B2(20~30mg/日)也可减低MetHb含量、改善发绀。这些药物对Ⅱ型b5R缺乏症神经系统病症无效。需注意,药物只能使发绀暂时减轻或消退,亚甲蓝虽见效快但不宜长期使用,可以用维生素C维持治疗。③合并G6PD缺乏的b5R缺乏症不能用亚甲蓝,可以选择维生素C,因为有报道亚甲蓝蓄积可引起呼吸困难、胸痛和溶血。
2.药物性高铁血红蛋白血症 无论是后天获得性还是在b5R缺陷基础上诱发加重以至危及生命,都应及时抢救。①立即停用可引起高铁血红蛋白血症的药物。②亚甲蓝1~2mg/kg静脉滴注或推注,注射时间不能短于5min。必要时1h内可重复1次。发绀消退后改为每天3~5mg/kg口服,维持治疗至血浆高铁血红蛋白浓度正常。用药后尿液呈蓝色,不良反应较轻,偶有膀胱刺激症状和肾结石。也可选择大剂量维生素C治疗。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










