



第二节 单基因遗传病
单基因遗传病是指受一对等位基因(主基因)影响而发生的疾病。单基因遗传病的遗传符合孟德尔定律,故又称为孟德尔式遗传病。
根据基因所在染色体的位置(常染色体或性染色体)和基因性质(显性或隐性)的不同,将单基因遗传病分为:常染色体显性遗传病、常染色体隐性遗传病、X连锁显性遗传病、X连锁隐性遗传病和Y连锁遗传病。据2007年世界统计数据显示,人类细胞核基因组决定的单基因遗传性状有17 654种,包括常染色体遗传的16 613种、X连锁遗传的985种、Y连锁遗传的56种。其中,与人类疾病相关的基因座有10 314个。目前记载的单基因遗传病中,一半以上的是常染色体显性遗传,约36%的是常染色体隐性遗传,约10%的为X连锁遗传。
一、系谱与系谱分析法
研究人类性状、疾病的遗传规律不能像研究动物、植物那样人为地进行杂交实验,而是通过观察这些性状或疾病在家系内分离或传递的结果进行推断,所以需要特殊的研究方法,即系谱分析法(pedigree analysis)。系谱分析法是临床上研究人类遗传性状和疾病最常用的方法。进行系谱分析既有助于判断患者是否有遗传病,又有助于区分该疾病是单基因遗传病、多基因遗传病还是线粒体遗传病,还可用于遗传咨询中个体患病风险的估计和基因定位中的连锁分析等。
所谓系谱(pedigree)是指从先证者入手,详细调查某种疾病在一个家族中的发病情况后,用规定的符号和一定的格式将调查结果绘制成患者与家族各成员相互关系的图解,又称为家系图。系谱中常用的符号如图5-8所示。
在系谱中,先证者是指该家族中第一个被医生或遗传研究者发现罹患某种遗传病的患者或具有某种性状的成员。系谱中不仅包括患病的个体,也包括家族中所有的健康成员。通过系谱可以对这个家系进行回顾性分析,以便确定所发现的某一疾病(或特定性状)在这个家系中是否有遗传因素的作用及其可能的遗传方式,从而为其他具有相同遗传病的家系或患者提供预防或诊治的依据。在系谱分析时,仅依据一个家族的系谱资料往往不能反映出该病遗传方式特点,通常需要将多个具有相同遗传性状或遗传病的家族系谱作综合分析(统计学分析),才能比较准确而可靠地作出判断。在调查和绘制系谱时还要注意:患者的年龄、病情、死亡原因、是否近亲婚配等;一个家族中检查的人数愈多愈好,大家族才能提供更多的信息,系谱一般要求有三代以上的成员情况;调查时要深入实地察看、查询,多收集资料进行综合分析,以确保资料准确无误;系谱中不能表达的内容应记录在病历内备查。
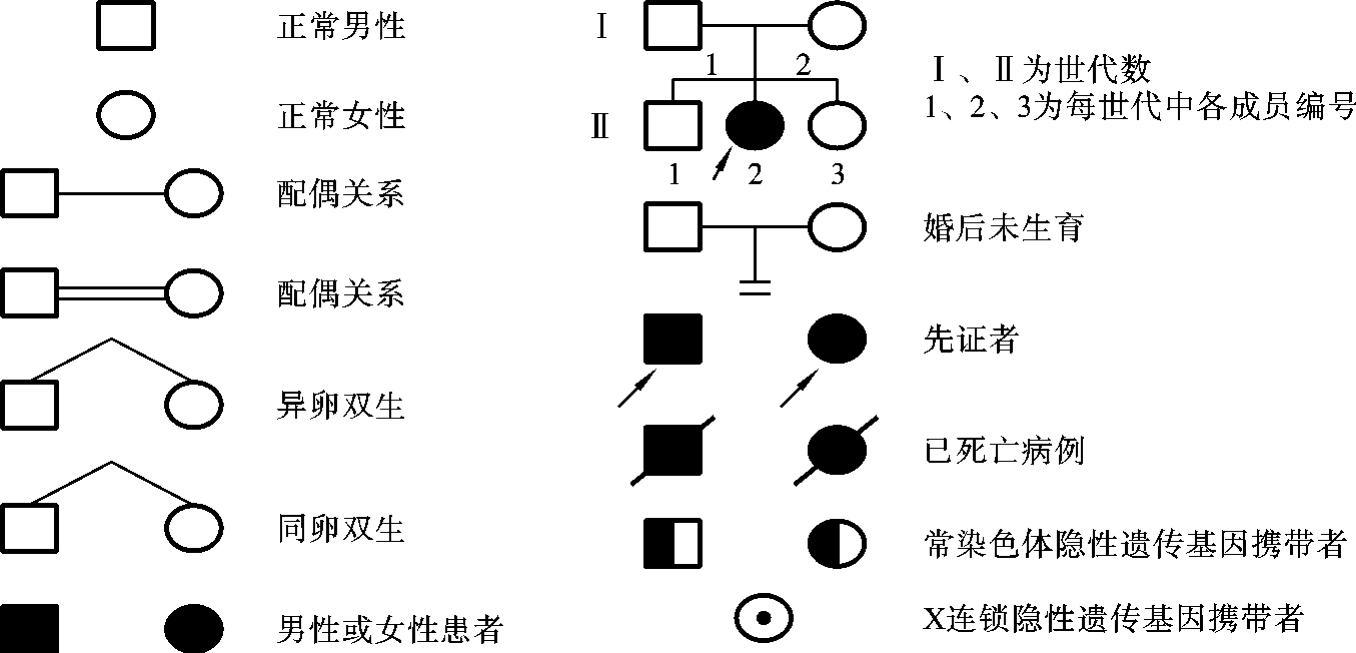
图5-8 系谱中常用的符号
二、常染色体显性遗传
控制某种性状或疾病的基因位于常染色体(第1~22号染色体)上,而且致病基因的性质是显性的,这种遗传方式就称为常染色体显性遗传(autosomal dominance inheritance,AD),由此引起的疾病称为常染色体显性遗传病。
在常染色体显性遗传病中,假定用A表示显性致病基因,用a表示相对应的隐性正常基因,则基因型为AA和Aa的个体患病,基因型为aa的个体正常。人类的致病基因最初都是由正常基因突变而来的。基因突变的频率是很低的,每代基因突变率为10-6~10-4,所以对于常染色体显性遗传病来说,患者的基因型绝大多数为杂合的基因型(Aa),纯合的基因型(AA)是致死的,在临床上比较少见。且在大多数病例中,纯合子患者的病情比杂合子患者的病情严重得多。
如果常染色体显性遗传病患者(Aa)与正常人(aa)结婚,婚后所生子女中约有1/2是患病个体,约有1/2为正常人。而且这样一对夫妇,他们每生育一次,其子女都有1/2的患病风险(图5-9)。
由于基因表达受到多种复杂的内、外环境因素的影响,杂合子(Aa)有可能出现不同的表现形式,因此将常染色体显性遗传又分为以下几种亚型。
(一)完全显性遗传
在常染色体显性遗传疾病中,杂合子(Aa)患者与显性纯合子(AA)患者的表现型完全相同,临床症状并无区别,称为完全显性遗传(complete dominance inheritance)。在杂合子(Aa)中,显性遗传基因A的作用完全表现出来,隐性基因a的作用完全被掩盖,从而使杂合子(Aa)表现出与显性纯合子(AA)完全相同的症状。
例如,短指症是常染色体完全显性遗传病的典型例子,它的主要症状是患者由于指骨短小或缺如,致使手指变短。已知短指对正常指为显性性状,如果用A表示短指基因,a表示正常指基因,基因型AA和Aa的个体都是短指症患者,临床表现相同,但临床上所见到的短指症患者的基因型绝大多数为杂合子(Aa),基因型(aa)的个体表现为正常指。
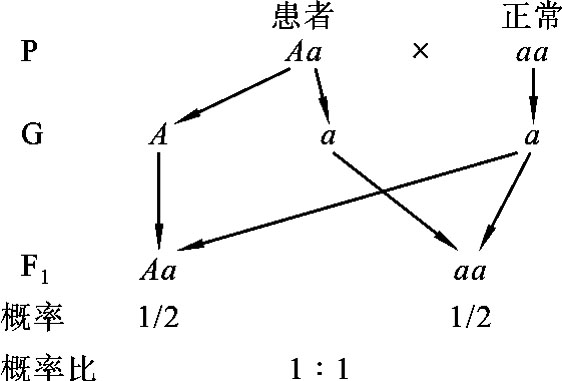
图5-9 常染色体显性遗传病患者与正常人婚配图解
当短指症患者(Aa)与正常指个体(aa)婚配,按孟德尔分离定律计算,其所生子女中约有1/2的是短指症患者,约有1/2的是正常指个体。也可以说,这对夫妇每生一个孩子都有1/2的可能是短指症患儿。图5-10是一例短指症家族系谱。
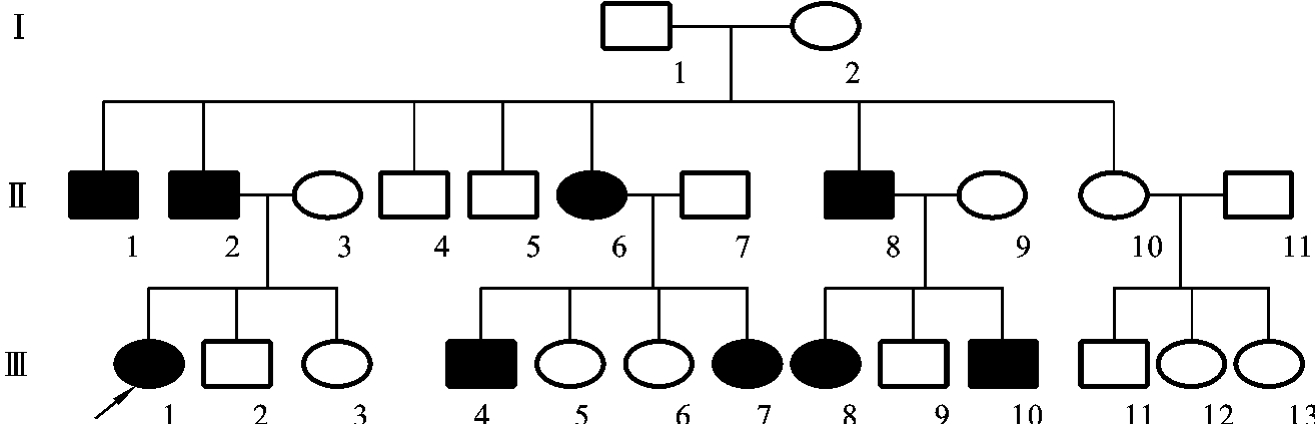
图5-10 一例短指症家族的系谱
并指症Ⅰ型也是完全显性遗传病,可伴有手部或其他部位畸形,常在双侧同时发生,呈对称性。以发生在中指和环指间者最多,有时可有3个、4个或全部手指并指。并指程度深浅不一,有的仅在指根部并指,表现为指蹼短浅;有的为全指并指,相邻手指完全连在一起(图5-11)。
图5-12显示的是一例并指症Ⅰ型家族的系谱。从以上典型的病例中可以看出,常染色体完全显性遗传具有如下特点。①由于致病基因位于常染色体上,因而致病基因的遗传与性别无关,即在系谱中男、女发病机会均等。②患者的双亲之一必有一方是患者,且患者绝大多数是杂合子,患者的同胞中约有1/2的为患者,患者的子女中约有1/2的为患者,也就是说,患者婚后每生育一次都有50%的概率生出该病患儿。在小家系中同胞的发病比例不一定能准确反映出来,但如果把相同病种、婚配方式相同的小家系总计起来分析,就可以看到近似的发病比例。③系谱中可见连续传递,即系谱中每代都可能出现患者。④双亲无病时,子女一般不会发病(除非发生新的基因突变或一些不规则显性遗传)。
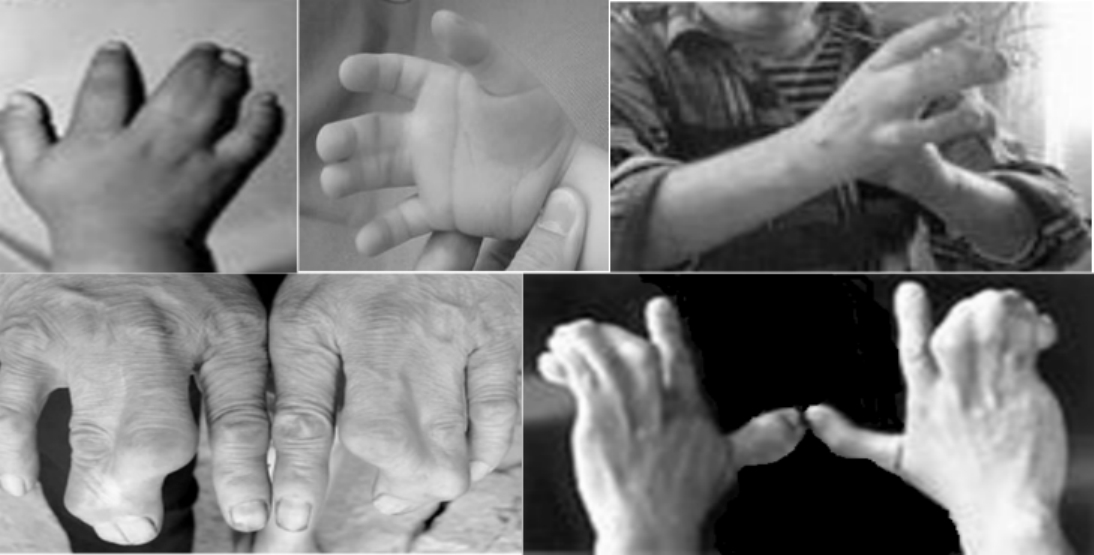
图5-11 并指症Ⅰ型患者的手部
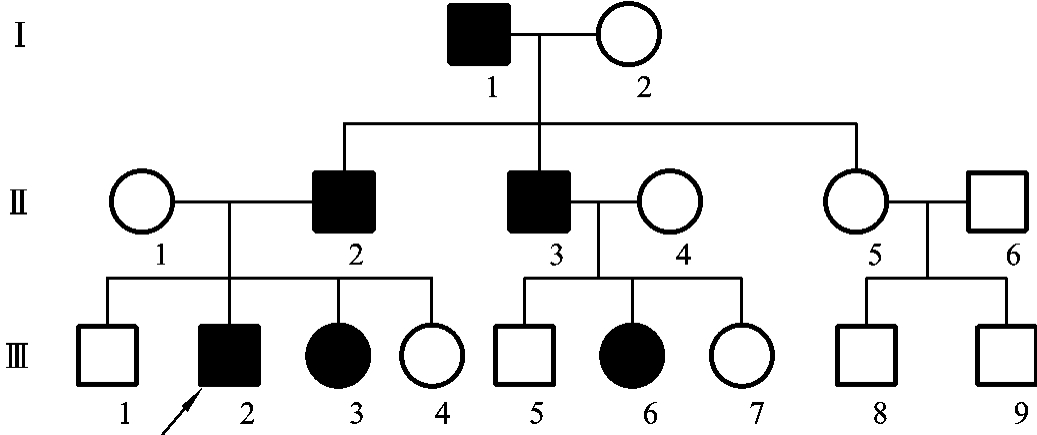
图5-12 一例并指症Ⅰ型家族的系谱
(二)不完全显性遗传
在常染色体显性遗传病中,杂合子(Aa)的表现型介于显性纯合子(AA)与隐性纯合子(aa)的表现型之间,称为不完全显性遗传(incomplete dominance inheritance)或半显性遗传(semi-dominance inheritance)。在杂合子(Aa)中,隐性基因a的作用也得到一定程度的表现,所以,在不完全显性遗传病中,杂合子(Aa)为轻型患者,纯合子(AA)为重型患者。两个杂合子(Aa)婚配后,后代的表型比例不是3∶1,而是1∶2∶1,如β-珠蛋白生成障碍性贫血、软骨发育不全症、家族性高胆固醇血症等。
β-珠蛋白生成障碍性贫血也称β-地中海贫血,在中国南方地区的人群中致病基因携带率达到10%以上,是由于血红蛋白β链合成障碍而造成的贫血。不同基因的个体,由于血红蛋白β链合成受到不同程度的影响,因而在临床上会出现不同的病情。①显性纯合子(βThβTh)是重型患者,不能合成或只能合成很少量的血红蛋白β链,因此患者在出生后几个月内便出现严重的进行性贫血,常靠输血维持生命,多在婴幼儿期夭折;②杂合子(βThβth)是轻型患者,血红蛋白β链合成部分受抑制,所以临床症状较轻,只表现轻度或中度贫血,一般可生活至成年;③隐性纯合子(βthβth)是正常人,血红蛋白β链合成正常(图5-13)。
软骨发育不全症又称胎儿型软骨营养障碍、软骨营养障碍性侏儒等,是一种由于软骨内骨化缺陷导致的先天性发育异常。本病纯合子(AA)患者病情严重,多在胎儿期或新生儿期死亡,而杂合子(Aa)患者在出生时即有体态异常:四肢粗短(侏儒),手指平齐,下肢向内弯曲,胸椎后突,腰椎前突,以后者更为明显,头颅增大(图5-14)。有的患者有轻度脑积水,颅穹窿及前额突出。大部分患者智力发展正常,牙齿好,肌力强,性功能正常。患者婴儿期如未夭折,成年后可以胜任各种工作,预后良好。本病主要是由于长骨骨骺端软骨细胞形成及骨化发生障碍,影响了骨的生长所致。致病基因已定位于4p16.3。
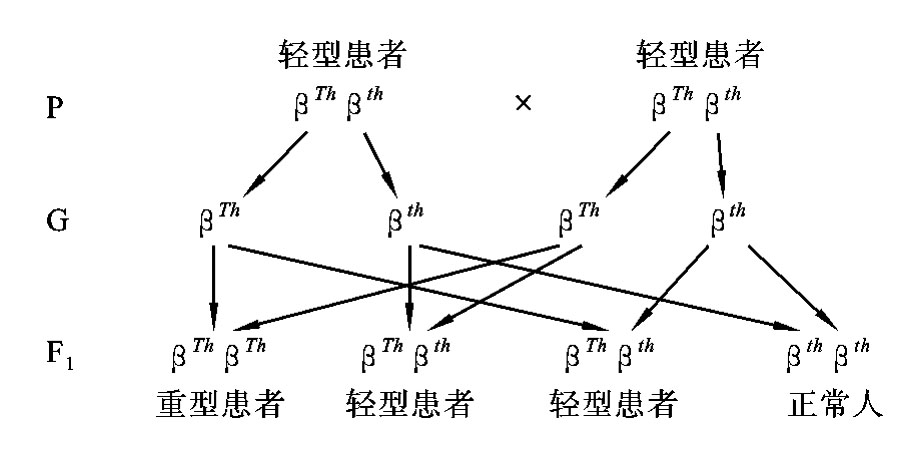
图5-13 两个轻型β-珠蛋白生成障碍性贫血患者婚配图解

图5-14 软骨发育不全症患者
(三)共显性遗传
一对等位基因之间彼此没有显性和隐性的区别,在杂合状态时,两者的作用都完全表现出来,这种遗传称为共显性遗传(codominance inheritance)。
人类血型系统有二十多种,其中,ABO血型和MN血型遗传就是共显性遗传典型的例子。ABO血型是由红细胞表面抗原决定的,红细胞表面有A抗原,血清中有β抗体者为A血型;红细胞表面有B抗原,血清中有α抗体者为B血型;红细胞表面有A、B抗原时,血清中无抗体者为AB血型;红细胞表面无A、B抗原,而血清中有α和β两种抗体时为O血型。
ABO血型是由一组复等位基因(multiple alleles)决定的,它们分别是IA、IB、i,这三种基因位于第9号染色体长臂的同一位点,互为等位基因,但对于每个人来说只能具有其中的两个基因。像这种在一对同源染色体的某一特定位点上有三种或三种以上的等位基因称为复等位基因,它是基因突变多向性的表现。IA决定红细胞表面有A抗原,IB决定红细胞表面有B抗原,i决定红细胞表面既没有A抗原又没有B抗原。IA和IB对i都是显性的;IA和IB之间无显性与隐性之分,而表现为共显性。因此,基因型IAIA、IAi表现为A血型,基因型IBIB、IBi表现为B血型,基因型ii表现为O血型,而基因型IAIB则表现为AB血型。
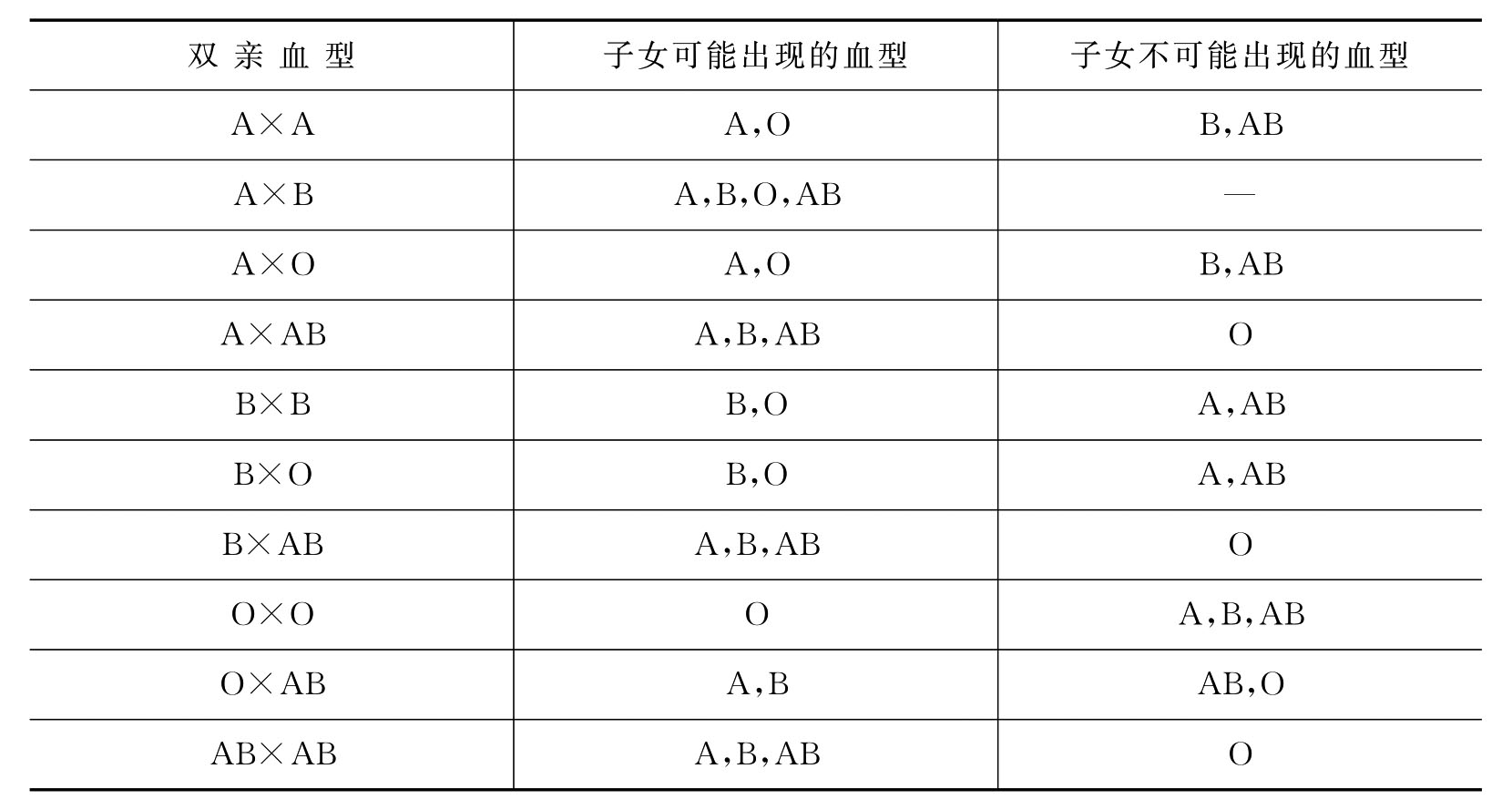
根据分离定律的原理,如果知道了双亲的血型,就可以推断出子女中可能出现什么血型或不可能出现什么血型(表5-1)。这在法医学的亲权鉴定上有一定的意义。
表5-1 双亲血型和子女血型的遗传关系
例如,如果父母双亲分别是AB血型或O血型,子女可能是A血型或B血型,不可能是AB血型和O血型(图5-15)。
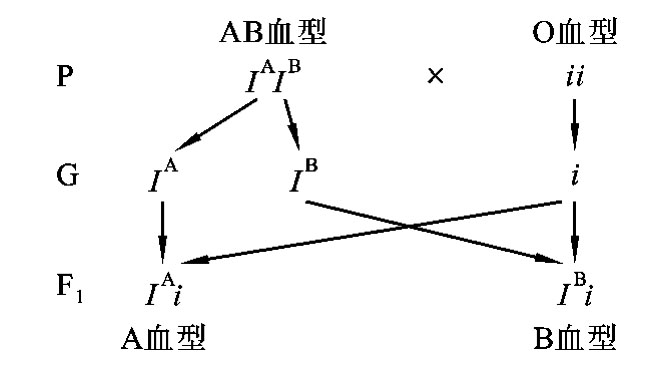
图5-15 AB血型个体与O血型个体婚配图解
知识链接
ABO血型不合
母婴ABO血型不合易引起新生儿溶血症,原因是在母亲妊娠期间,胎儿红细胞可通过胎盘进入母体,刺激母体产生新的血型抗体,该抗体又通过胎盘进入胎儿体内,与红细胞发生抗原抗体反应,可引起红细胞破裂。因个体差异,母体产生的血型抗体量及进入胎儿体内的量不同,当胎儿体内的抗体达到一定量时,导致较多红细胞破裂,表现为新生儿溶血症。如母亲为O血型,父亲为A血型、B血型或AB血型,如果胎儿为A血型或B血型时,母亲的抗A或抗B抗体通过胎盘进入胎儿血液循环,可发生免疫性溶血反应导致新生儿溶血性黄疸,这种情况在我国比较常见,北方地区约半数新生儿黄疸是由此引起的。引起新生儿溶血性黄疸的因素较多,仅仅查出母婴ABO血型不合还不能确诊,尚需检查新生儿血中是否有抗A或抗B抗体,母血中抗A或抗B的抗体浓度也应相当高。ABO血型不合导致溶血症状一般较轻,引起胆红素脑病致死者较少见。
(四)不规则显性遗传
在有些常染色体显性遗传病中,杂合子(Aa)由于所处的遗传背景和环境因素的影响,使显性基因的作用没能表达出来,或者表达的程度有差异,使显性性状的传递不规则,这种遗传现象称为不规则显性遗传(irregular dominance inheritance),如多指症、Marfan综合征、成骨发育不全综合征、Ⅰ型神经纤维瘤等。
如在多指症中,有些杂合子(Aa)个体携带有显性致病基因(A),不一定表现有疾病,但显性致病基因依然可以向后代传递,使后代中出现该病的患儿,因此在系谱中可以出现隔代遗传的现象。
图5-16是一例多指症家族的系谱,在该系谱中,先证者Ⅱ2的3个子女中一个正常,2个患有多指症,这证明该先证者是杂合子(Aa),而他的父母Ⅰ3和Ⅰ4都正常,而其伯父Ⅰ1是多指症患者。由此可见,他的父亲Ⅰ3可能是杂合子(Aa),由于其遗传背景和环境因素的影响,而使显性致病基因(A)的作用未能显示出来,所以父亲Ⅰ3的手指正常,但并不影响其将致病基因传给后代,使后代出现患者,从而出现了隔代遗传的现象。
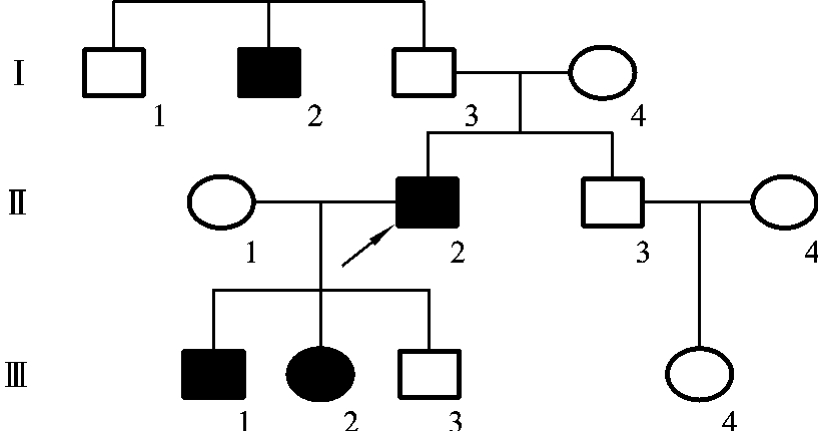
图5-16 一例多指症家族的系谱
形成上述这种现象的最常见的原因之一是外显率降低。所谓外显率是指具有一定基因型的个体在特定环境中形成相应表现型的比例,一般用百分率(%)来表示。外显率等于100%者为完全外显,外显率低于100%者为不完全外显或外显不全。如有20名携带有致病基因A的杂合子,只有16名形成了与显性基因相应的症状,另外4人未出现相应的症状,则该杂合子(Aa)的外显率为16/20×100%=80%。
另外,在多指症的另一些杂合子(Aa)个体中,显性基因A的作用虽然表现出相应的症状,但不同个体间症状表现出的轻重程度有所不同,如有的多指数目不一,有的多指长短不等(图5-17)。这种杂合子(Aa)因某种原因而导致的个体间表现程度(轻重)的差异,一般用表现度(expressivity)来表示。

图5-17 多指症患者的手部
Marfan综合征(Marfan syndrome)也是一种比较常见的不规则显性遗传病。患者的主要患病器官为骨骼、心血管系统和眼。临床表现为:患者的身体瘦高、四肢细长,两臂伸长的长度大于身高,躯体上半部(头顶到耻骨联合)与下半部(耻骨联合到脚底)的比例降低,手指如蜘蛛样,颅骨细长,硬腭高拱,常见鸡胸或漏斗胸,常伴有韧带松弛及脊柱侧凸。眼部的典型损害为晶状体脱位,也可出现高度近视眼、视网膜剥离等。该病60%~80%的患者有心血管系统疾病,最常见是二尖瓣的功能障碍,二尖瓣腱索破裂和主动脉瘤破裂可引起过早死亡。本综合征的重型患者可有骨骼、眼和心血管系统的严重损害;轻型患者则只有各器官不同程度的损伤或只有骨骼和眼的异常或骨骼和心血管系统的异常。同一家族中的不同患者可有不同器官的不同程度损害,表现为不规则显性遗传(图5-18)。
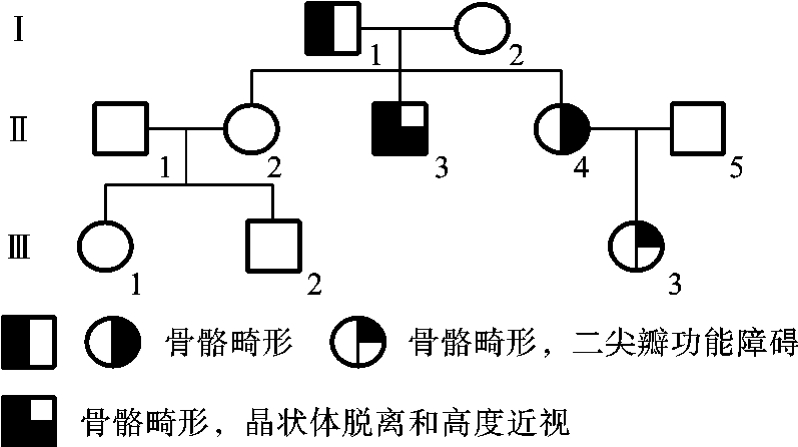
图5-18 一例Marfan综合征家族的系谱
成骨发育不全综合征(osteogenesis imperfecta syndrome)也是一种典型的表现度有个体差异的例子,主要临床表现为骨折、蓝色巩膜和进行性传导性耳聋。在同一家系中,基因型相同的个体由于个体之间表现度的不同而导致其临床表现也不同。重型患者可有早发和频发的骨折,脊柱侧凸,2/3的患者有白色巩膜,而1/3的患者有蓝色巩膜,有耳聋和牙本质发育不全。轻型患者有骨折或蓝色巩膜,无脊柱侧凸和耳聋。
值得注意的是外显率与表现度是两个不同的概念,切不可混淆。两者根本的区别在于前者阐明了基因表达与否,是“质”的概念,而后者要说明的是在表达前提下的表现程度的差异,是“量”的概念。
(五)延迟显性遗传
在常染色体显性遗传中,杂合子(Aa)在生命的早期致病基因并不表达,或虽然表达但尚不足以引起明显的临床表现,只有在达到一定的年龄后才表现出疾病,这种遗传方式称为延迟显性遗传(delayed dominance inheritance)。
亨丁顿(Huntington)舞蹈症又称慢性进行性舞蹈症,是一种常染色体显性遗传病。患者的子女中半数者可患病,男、女患病机会均等,多在30~40岁开始发病,但也有10余岁或60岁以后才发病的病例。临床表现为不自主舞蹈样动作及进行性痴呆。脑体积明显缩小,重量小于1 000g,最突出的是两侧尾状核和壳核的萎缩,以致侧脑室明显扩张(图5-19)。大脑皮质特别是额叶、顶叶萎缩显著,白质也减少。镜下可见尾状核和壳核中选择性小神经细胞丢失,伴星形胶质细胞增生和胶质纤维化,类似的病变可见于丘脑腹侧核和黑质。

图5-19 亨丁顿舞蹈症患者及其脑部
本病呈进行性发展,病程多为10~15年,最后死于并发症。亨丁顿舞蹈症的致病基因已定位于4p16.3。该基因5′端有(CAG)n三核苷酸重复序列。正常人重复9~34次,平均20次;该病患者重复37~100次,平均46次。
脊髓小脑性共济失调Ⅰ型也是一种常染色体显性遗传病,常于30~40岁发病,但也有14岁之前或73岁以后发病的。患者有小脑萎缩,脑桥和橄榄体变性,脊髓小脑束萎缩。患者步态不稳,行走困难,上肢动作笨拙,语言不清,吞咽困难,有摇头和舞蹈样动作。此外可有眼外肌麻痹、眼震颤、腱反射亢进。该病的致病基因定位于6p23,致病基因的产物称为共济失调蛋白。其基本缺陷在于外显子中的三核苷酸(CAG)n的重复扩展。正常人的CAG重复25~39次,该病患者的CAG重复51~58次。
家族性多发性结肠息肉也是延迟显性遗传病。该病患者的结肠壁上有许多大小不等的息肉,临床主要症状为便血并伴有黏液。35岁前后,患者的结肠息肉可恶化成结肠癌。
(六)从性遗传
从性遗传(sex-influenced inheritance)是指杂合子(Aa)的表达受到性别的影响,在某一性别表现出相应表型,在另一性别则不表现出相应的性状。例如,秃顶(又名男性秃顶)为常染色体显性遗传病,表现为成年男性头前部至头顶头发的慢性脱落,枕部及两侧颞部仍保留正常头发,男性杂合子有的表现秃顶,女性杂合子则不表现秃顶,但可以传递给后代,也就是说,在她的后代中有1/2的男性可出现秃顶。这种表达上的差异可能与雄激素的作用有关。
再如原发性的血色素沉着症,也为常染色体显性遗传病,主要表现为皮肤色素沉着、肝硬化、糖尿病三联综合征,人群中男性的发病率高于女性的。女性由于月经、流产或妊娠等生理或病理失血导致铁质缺失,减轻了铁质的沉积,故不易表现出症状。
三、常染色体隐性遗传
控制某种性状或疾病的基因位于常染色体上,而且致病基因的性质是隐性的,这种遗传方式称为常染色体隐性遗传(autosomal recessive inheritance,AR)。由常染色体上的隐性致病基因引起的疾病称为常染色体隐性遗传病。目前已被确认的常染色体隐性遗传病有1 730种,如白化病、苯丙酮尿症Ⅰ型、尿黑酸尿症、先天性聋哑、高度近视、半乳糖血症、肝豆状核变性、镰形细胞贫血症等。先天性代谢病多数为常染色体隐性遗传。
在常染色体隐性遗传的等位基因B和b中,b为突变致病基因,个体只有在基因型为bb时才表现为疾病,而杂合子(Bb)虽然带有一个致病基因,但隐性致病基因(b)的作用会被显性正常基因(B)掩盖,因此杂合子(Bb)的表型与正常人(BB)相同,但却可以将隐性致病基因遗传给后代。这种带有致病的遗传物质(基因)但表型正常,并能将遗传物质传递给后代的个体称为携带者(carrier)。最常见的婚配类型是两个携带者(Aa×Aa)婚配(图5-20)。
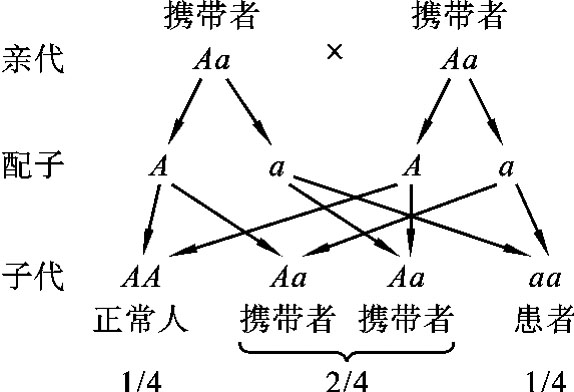
图5-20 两名白化病携带者婚配图解
白化病(albinism)是较常见的常染色体隐性遗传病。患者的皮肤、毛发呈白色,虹膜呈淡灰色,畏光,眼球震颤。由于患者体内编码酪氨酸酶的基因发生突变,导致酪氨酸酶缺陷,不能产生黑色素。该病的致病基因位于15q11—q12。
苯丙酮尿症Ⅰ型(phenylketonuria,PKUⅠ),是一种遗传性酶病,在我国的发生率为1/16 500。患儿出生时正常,毛发淡黄,皮肤白皙,虹膜呈黄色,尿有鼠臭味或霉臭味。出生后3~4个月,患儿出现智力发育障碍,肌张力高,常有痉挛发作,行走时步态不稳。约有1/2的患病胎儿发生早期流产,1/2的患儿生长迟缓,有小头并有严重的智力低下。
本病主要是由于苯丙氨酸羟化酶(PHA)基因缺陷,引起苯丙氨酸羟化酶遗传性缺陷,导致苯丙氨酸不能转变为酪氨酸,结果在血清中积累。过多的苯丙氨酸进入旁路代谢,生成苯丙酮酸、苯乳酸和苯乙酸,堆积于人体内而导致疾病。PHA基因已定位于12q12.1。
再如镰形细胞贫血症患者因β珠蛋白基因的第6位谷氨酸变为缬氨酸,导致红细胞变成镰刀状而致病。从杂合子(HbAHbS)患者的症状上来看,是常染色体隐性遗传病,但杂合子(HbAHbS)患者的血细胞在显微镜下看,既有正常细胞,还有镰刀状的细胞,属于共显性遗传。图5-21是正常人与镰形细胞贫血症患者纯合子(HbS HbS)、镰形细胞贫血症患者杂合子(HbAHbS)与镰形细胞贫血症患者纯合子(HbS HbS)之间婚配的图解。
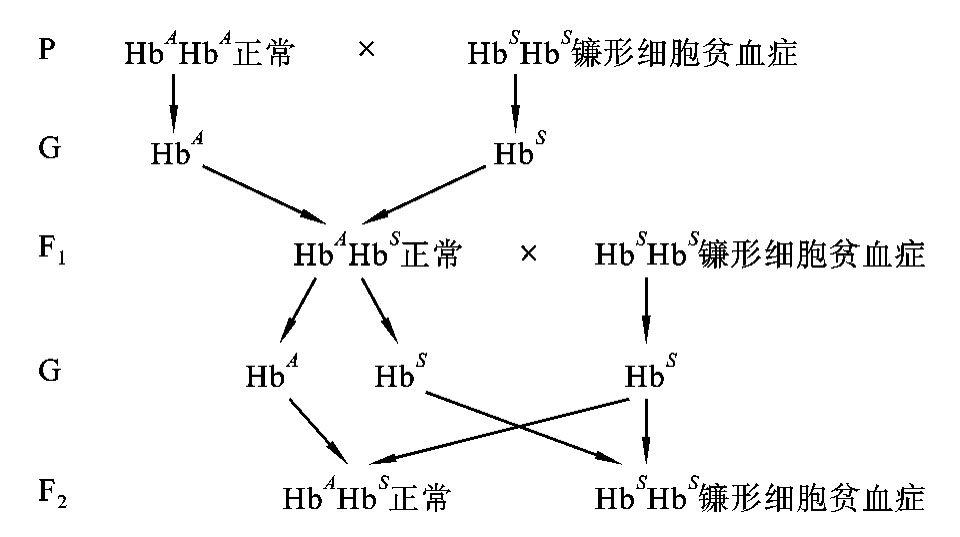
图5-21 镰形细胞贫血症患者与正常人之间的婚配图
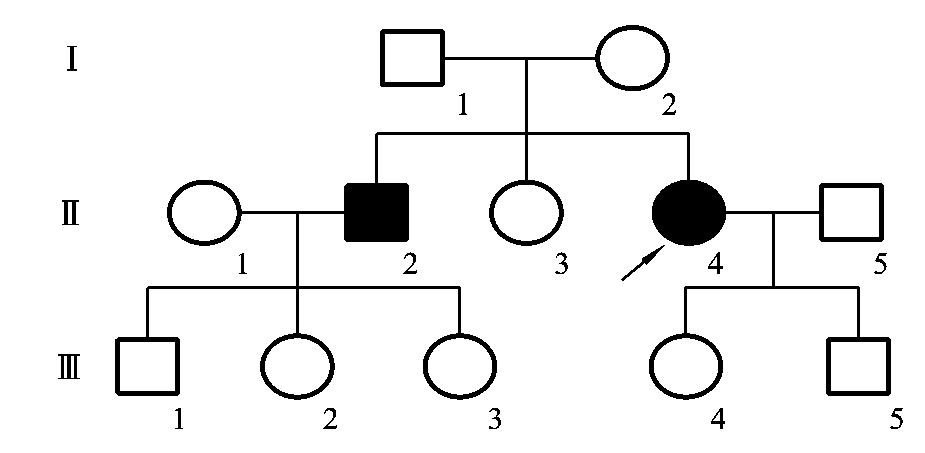
图5-22 白化病患者的系谱
图5-22是一例白化病患者的系谱。从该系谱中可以看到常染色体隐性遗传的系谱特点:①患者的双亲往往表型为正常,但都是致病基因的携带者;②患者的同胞患同样疾病的可能性为1/4,表型正常的同胞中有2/3的可能性为携带者;③系谱中看不到连续传递,系谱中的患者往往是散发的;④由于致病基因位于常染色体上,故男、女患病机会相等;⑤近亲婚配时,后代发病概率明显增高。
值得注意的是,常染色体隐性遗传病患者的同胞患同样疾病的可能性在理论上为1/4,但临床上观察的结果往往大于1/4,这是由于小家系中同胞数较少,有时看不到准确的发病比例,如果将相同婚配类型的小家系合并起来分析,就会看到近似的发病比例。
所谓近亲婚配是指在3~5代内有共同祖先的男、女进行婚配。由于近亲个体可能携带有共同祖先传递下来的相同致病基因,所以亲缘关系越近则携带有相同致病基因的可能性就越高。近亲婚配时,子女成为隐性纯合子的机会比随机婚配时要高许多倍,所以近亲婚配子女发病的风险会增高。
历史上,有些国家和地区曾鼓励近亲婚配,不过,现在大多数国家都已禁止。我国《婚姻法》中有直系血亲和三代以内的旁系血亲禁止结婚的规定,所以我国近亲婚配的比率大大降低。可是在一些偏僻、落后的农村或山区以及一些少数民族地区,还存在着近亲婚配的现象。
在遗传学上,亲缘关系的远近通常用亲缘系数来表示。亲缘系数(relationship coefficient,r)是指具有共同祖先的个体,在某一基因位点上携带有相同基因的概率,或者说,两个个体(如表兄妹)之间具有相同基因的可能性。亲缘关系越近,亲缘系数越大。亲缘关系与亲缘系数的关系见表5-2。
表5-2 亲缘关系与亲缘系数的关系

可以从两方面理解亲缘系数:①两个个体之间基因总体上有多少是相同的:如父子、母子、同胞之间有1/2的基因是相同的;祖孙之间、姑侄之间有1/4的基因是相同的;表兄妹之间有1/8的基因是相同的等。②从某一对等位基因来讲,两个个体在同一个基因位点上共同具有同一个基因的可能性。在亲缘系数中这个基因相同更侧重于从功能上的相同。假如一对有显、隐性区别的等位基因A和a,有亲缘关系的两个个体都含有一个致病基因a,这个致病基因可能来自于一个共同祖先的其中一方,或来自于共同祖先的双方。
假如半乳糖血症在人群中携带者的概率是1/150,两个携带者随机婚配的概率为1/150×1/150=1/22 500,随机婚配每生育一次可能生出患儿的风险是1/22 500×1/4=1/90 000。如果是表兄妹婚配,设表兄为群体携带者,即携带致病基因的概率为1/150,则表妹有1/8的可能性携带有与表兄相同的致病基因,所以他们婚配生出患儿的概率为1/150×1/8×1/4=1/4 800。即表兄妹婚配后生出半乳糖血症患儿的风险要比随机婚配增高约19倍,可见近亲婚配的危害性。
四、X连锁遗传
控制某一性状或疾病的基因位于X染色体上,这些基因将随着X染色体的传递而传递,这种遗传方式称为X连锁遗传(X-linked inheritance)。在X连锁遗传中,根据致病基因性质的不同又将其分为X连锁显性遗传和X连锁隐性遗传两类。目前已被确认的X连锁遗传病有412种。
在X连锁遗传中有两大特点:一是男性为半合子,由于男性只有一条X染色体,其X染色体上的基因在Y染色体上缺少与之对应的等位基因,因此男性只有成对基因中的一个成员,故称为半合子(hemizygote),只要X染色体上有致病基因,都可以表现出相应的症状;二是存在交叉遗传现象,所谓交叉遗传(criss-cross inheritance),是指在X连锁遗传中男性的X染色体上的致病基因只能来源于自己的母亲,将来只能传给自己的女儿,不存在由男性向男性传递的现象。
(一)X连锁显性遗传
由位于X染色体上的显性致病基因所控制的性状或疾病的遗传,称为X连锁显性遗传(X-linked dominant inheritance,XD)。由X染色体上的显性致病基因引起的疾病称为X连锁显性遗传病,临床上常见的有抗维生素D性佝偻病、遗传性肾炎、色素失调症、高氨血症Ⅰ型、口面指综合征和Albright遗传性骨营养不良等。
在X连锁显性遗传病中,假定显性致病基因为A,隐性正常基因为a,则男性的基因型有两种:XAY(患病)和XaY(正常)。女性的基因型有三种:XAXA(患病)、XAXa(患病)和XaXa(正常)。由于女性有两条X染色体,只要其中任何一条X染色体带有致病基因就会患病,所以女性的发病率高于男性的发病率。在临床上见到的女性患者绝大多数是杂合子(XAXa),很少见到女性纯合子患者,且女性患者的病情往往较轻。这是因为常见婚配类型为:女性杂合子患者(XAXa)×正常男性(XaY)(图5-23)或男性患者(XAY)×正常女性(XaXa)(图5-24),很少见到女性纯合子患者(XAXA)×男性患者(XAY)的婚配类型。
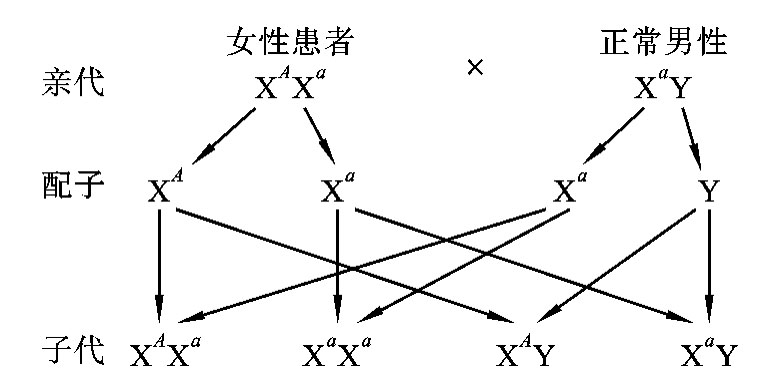
图5-23 女性患者与正常男性婚配图解
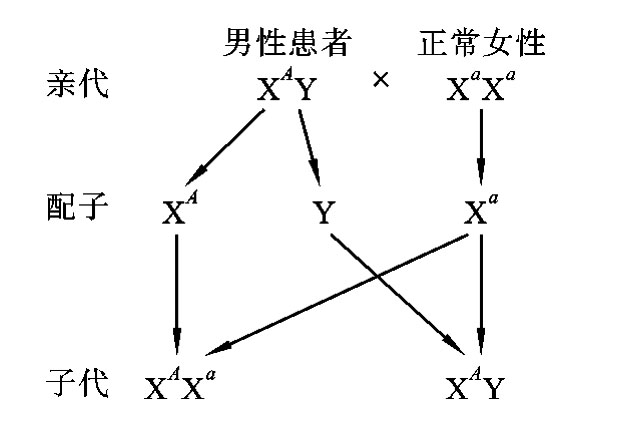
图5-24 男性患者与正常女性婚配图解
抗维生素D性佝偻病属于X连锁显性遗传病。致病基因位于Xp22.2—p22.1。患者由于肾小管对磷酸盐的重吸收和小肠对钙、磷的吸收障碍,造成尿磷增加、血磷降低、骨质钙化不全而引起佝偻病。患儿多于1岁左右开始发病,可出现O形腿、X形腿、鸡胸等骨骼发育畸形,以及多发性骨折、骨痛、不能行走和生长发育缓慢等临床表现(图5-25)。女性患者的病情较男性患者轻,少数只有低磷酸盐,或仅有骨骼异常。男性患者病情严重,下肢常出现畸形。这种佝偻病不仅会出现在婴儿期,在整个儿童期都存在,甚至在青春期仍在进展。对于这种佝偻病,采用常规剂量的维生素D治疗无效果,必须联合使用大剂量的维生素D和磷酸盐才能起到治疗效果,故本病称为抗维生素D性佝偻病。

图5-25 抗维生素D性佝偻病患者的外观及腿部骨骼X片
X连锁显性遗传的系谱特点:①系谱中女性患者多于男性患者,但女性患者的病情往往较男性患者的轻;②患者的双亲中有一方是患者,如果双亲都无病,子女一般不会患病(基因突变除外);③系谱中可见连续遗传现象;④有交叉遗传现象,即男性患者的后代中,女儿都将是患者,儿子都正常,女性患者的后代中子女各有1/2的患病风险(图5-26)。
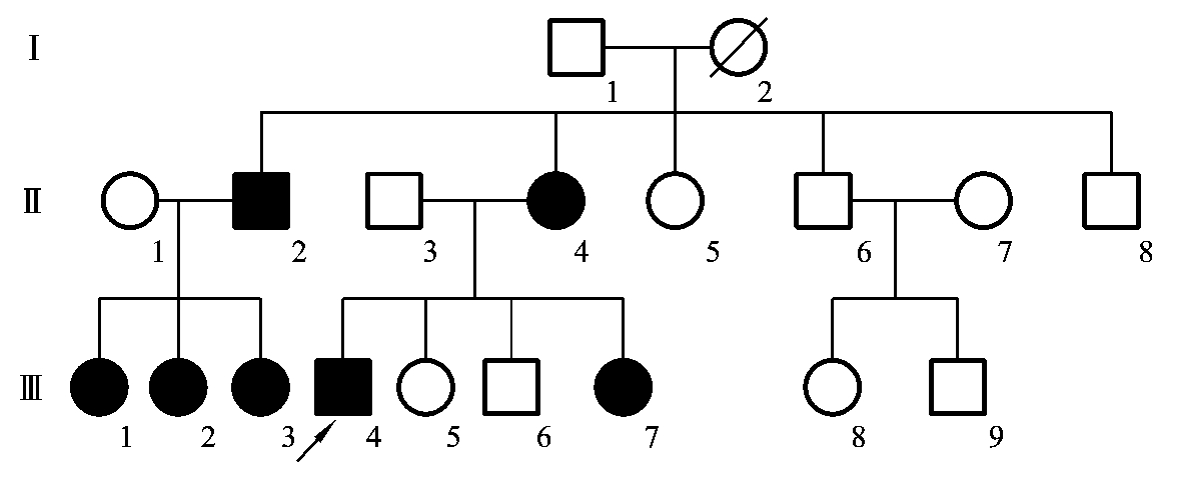
图5-26 一例抗维生素D性佝偻病家族的系谱
(二)X连锁隐性遗传
由位于X染色体上的隐性基因所控制的性状或疾病的遗传,称为X连锁隐性遗传(X-linked recessive inheritance,XR)。由X染色体上的隐性致病基因引起的疾病称为X连锁隐性遗传病,临床上常见有红绿色盲、甲型血友病、假肥大型肌营养不良和家族性低血色素贫血等。
在X连锁隐性遗传病中,致病基因为a,正常基因为A,则男性的基因型有两种:XAY(正常)和XaY(患者)。女性的基因型有三种:即XAXA(正常)、XAXa(携带者)和XaXa(患者)。当女性两条X染色体上的等位基因都是隐性纯合子(XaXa)时才表现为患者,当为杂合子(XAXa)时隐性基因控制的性状不表现出来。而男性只有一条X染色体,所以只要X染色体上有一个隐性致病基因(XaY)就会患病,所以男性的发病率高于女性的发病率。常见婚配类型:女性携带者(XAXa)×正常男性(XAY);女性携带者(XAXa)×男性患者(XaY)。
红绿色盲患者不能正确区分红色和绿色,该病由X染色体上两个紧密相连的隐性红色盲基因和绿色盲基因决定,一般把它们综合在一起考虑,称为红绿色盲基因。红绿色盲的发病率在男、女性别中差别很大,在我国男性红绿色盲的发病率约为7%,女性的发病率约为0.5%。
如果一个红绿色盲男性患者(XaY)与一个色觉正常女性(XAXA)婚配,子代中女儿色觉正常,但都是携带者,儿子都正常(图5-27)。男性患者的致病基因只传给女儿,不传给儿子。
如果红绿色盲女性携带者(XAXa)与色觉正常男性(XAY)婚配,子代中女儿全部色觉正常,但有1/2的是携带者,儿子中将有1/2的为患者(图5-28)。携带者母亲的致病基因可传给女儿,也可传给儿子,使儿子患病,故男性患者的致病基因只能从母亲那里传来。
如果红绿色盲女性携带者(XAXa)与男性色盲患者(XaY)婚配,子代中儿子将有1/2的为患者,1/2的正常;女儿中将有1/2的为患者,1/2的正常,但都是携带者(图5-29)。因此女性患者的父亲一定是患者,母亲若表型正常,则一定为携带者。
假肥大型肌营养不良主要发生于男孩,患儿的肌膜透过性增强,肌肉中的一些酶漏出至血液中,因此引起肌肉变性。
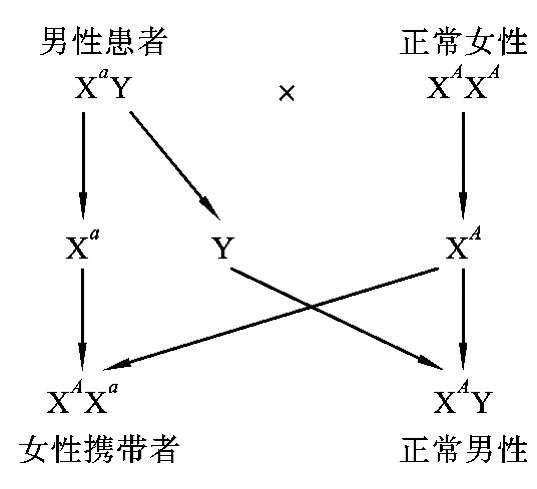
图5-27 红绿色盲男性患者与正常女性婚配
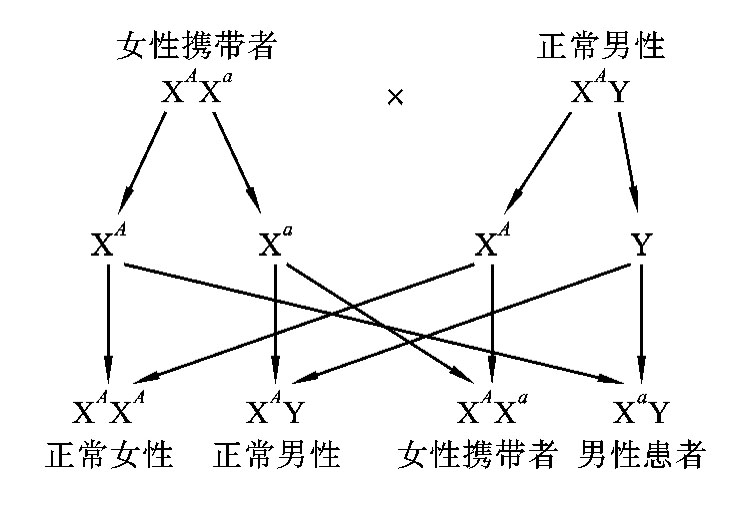
图5-28 红绿色盲女性携带者与正常男性婚配
Duchenne型肌营养不良(Duchenne muscular dystrophy,DMD)是假肥大型肌营养不良的主要类型,是严重致死性神经肌肉系统的XR病,其发病率为出生男婴的1/3 500。DMD患儿开始走路时就显现出肌肉无力,多在5~6岁时症状明显,表现为走路呈鸭型步态、上下楼困难,Gower征呈阳性,多数患者的腓肠肌为假性肥大(图5-30)。患儿的肌萎缩进行性加重,到10岁左右不能自主走路,一般20岁之前死于呼吸系统及循环系统衰竭,部分患儿伴有不同程度的智力低下。假肥大型肌营养不良的另一类型为Becker型肌营养不良(BMD),其发病率比DMD的明显低,但BMD发病较晚,一般10岁左右开始发病,临床表现与DMD的相似,但病程缓慢,症状较轻,往往可以生育后代。DMD和BMD都是由抗肌萎缩蛋白遗传性缺陷所致。
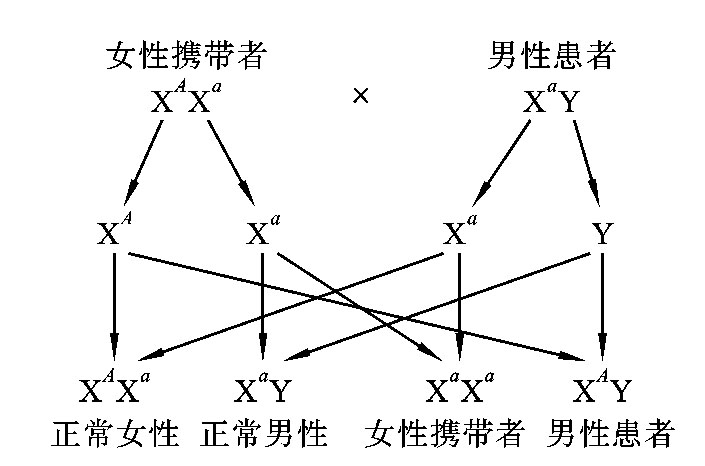
图5-29 红绿色盲女性携带者与男性患者婚配
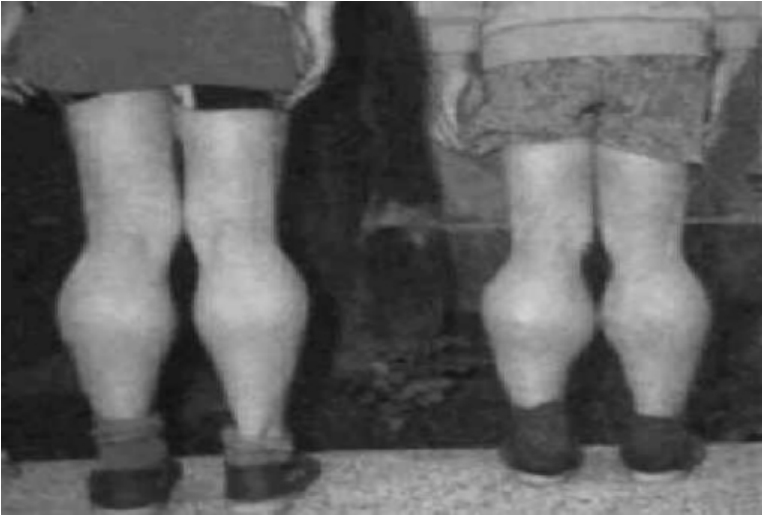
图5-30 假肥大型肌营养不良患者的腓肠肌假性肥大
DMD基因定位于Xp21.2—p21.3,是目前已知最大的致病基因,约占X染色体的1%,长约2 400kb,含79个外显子。
图5-31为一例假肥大型肌营养不良家族的系谱,该系谱反映了X连锁隐性遗传病系谱的特点:①人群中男性患者远多于女性患者,且系谱中往往只能见到男性患者;②有交叉遗传和隔代遗传现象;③双亲无病时,儿子可能发病,女儿则不会发病;儿子一旦发病,其母亲一定是致病基因的携带者;④男性患者的兄弟、外祖父、舅父、姨表兄弟、外甥及外孙等也可能是患者。
甲型血友病也是一种典型的X连锁隐性遗传病,致病基因定位于Xq28。患者由于缺乏凝血因子Ⅷ而导致凝血障碍,在皮下、肌肉内反复出血,可形成淤斑和血肿,在关节腔出血可导致关节畸形(图5-32),严重者可因颅内出血而死亡。
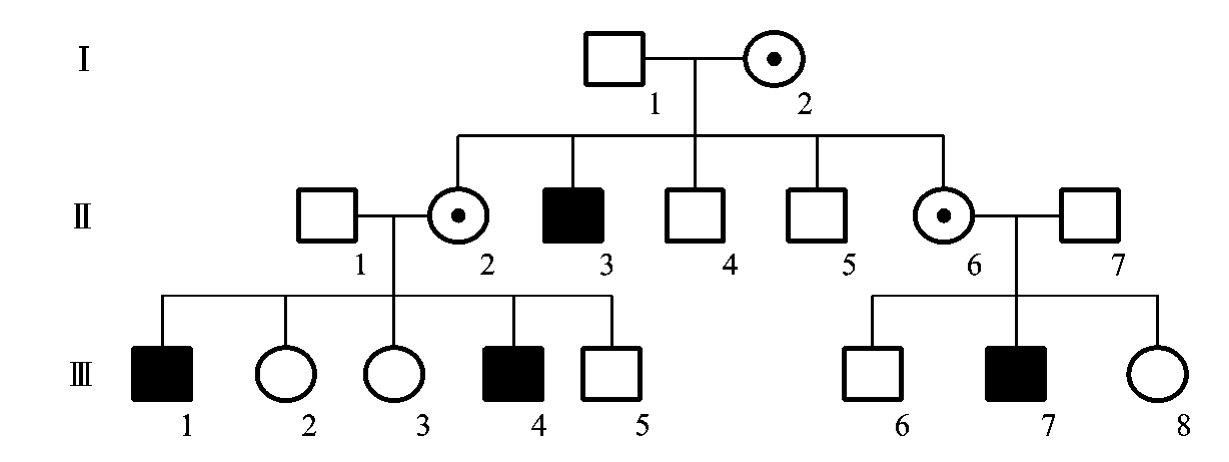
图5-31 一例假肥大型肌营养不良家族的系谱
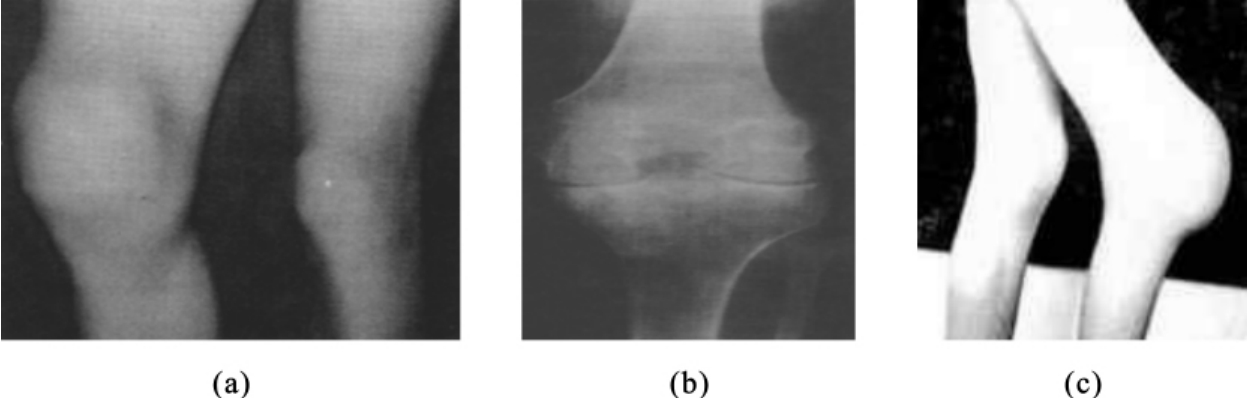
图5-32 甲型血友病患者的膝关节图片
五、Y连锁遗传
由位于Y染色体上的基因所控制的性状或疾病的遗传称为Y连锁遗传(Ylinked inheritance)。由Y染色体上的致病基因引起的疾病称为Y连锁遗传病。由于X染色体上缺少相应的等位基因,因此它必将随着Y染色体而传递,即只能从父亲传递给儿子,再由儿子传递给孙子,所以Y连锁遗传又称全男性遗传或限雄性遗传。据2002年统计,Y连锁致病基因有43个,目前已被确认的Y连锁遗传病有19种。
如外耳道多毛症是一种Y连锁遗传病,该家系中的全部男性个体在外耳道上可长出2~3cm长的成丛黑色硬毛,常伸出耳孔之外(图5-33)。该家系中的全部女性个体均无此症状(图5-34)。外耳道多毛症受Y染色体上的外耳道多毛基因控制。

图5-33 外耳道多毛症患者
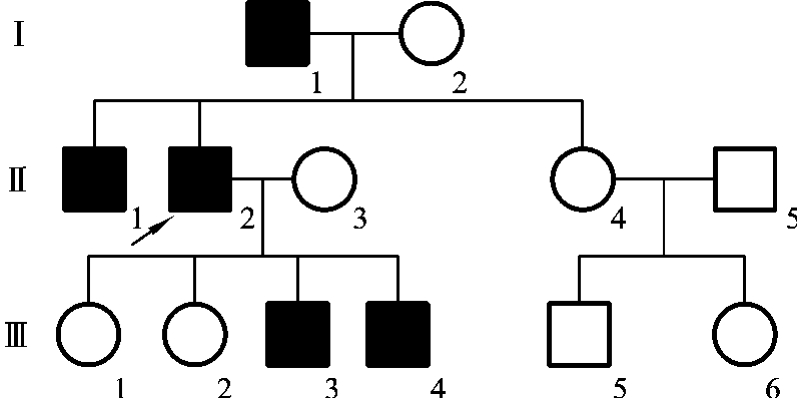
图5-34 一例外耳道多毛症家族的系谱
此外,目前较肯定的Y连锁基因还有睾丸决定基因、无精子基因、箭猪病基因、H-Y抗原基因等。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










