



2.常染色体隐性遗传

白化病小孩
常染色体隐性遗传病致病基因在常染色体上,其基因性状是隐性的,也就是说只有纯合子时才显示病状。此种遗传病父母双方均为致病基因携带者,因此往往见于近亲婚配者的子女。子代有1/4的概率患病,子女患病概率均等。许多遗传代谢异常的疾病,属常染色体隐性遗传病。按照“一个基因、一个酶”的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常。常见的常染色体隐性遗传病有溶酶体贮积症,如糖原贮积症、脂质贮积症、粘多糖贮积症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆状核变性及半乳糖血症等。
由于常染色体隐性遗传病是由位于常染色体上的隐性致病基因所引起的,所以具有以下特点:
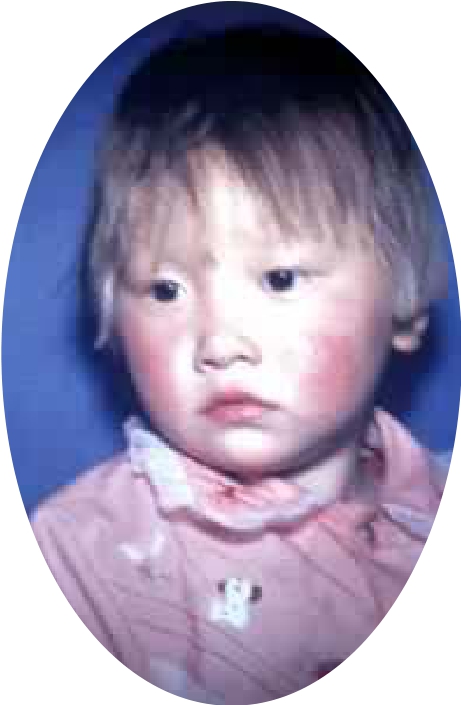
苯丙酮尿症患者
一是患者是致病基因的纯合体,虽然其父母不一定发病,但都是致病基因的携带者(杂合体)。
二是患者的兄弟姐妹中,约有1/4的人患病,男女发病的几率均等。
三是家族中不出现连续几代遗传,患者的双亲、远祖及旁系亲属中一般没有同样的病人。
四是近亲结婚时,子代的发病率明显升高。
(1)先天性葡萄糖、半乳糖吸收不良症

富含半乳糖的奶和鸡蛋
患儿呈现腹泻,为水样便,与尿很相似。腹泻的发生和程度与喂给糖的时间及量有关,给食后24小时出现腹泻,进食越多越严重,但患儿进食量却又很大,所以体重很快下降。继而呈现脱水、失重、营养不良。患儿只要不以果糖作为主要糖类来源,就会出现腹泻,故要终身限制食用葡萄糖及半乳糖。但随年龄增长对葡萄糖和半乳糖有所耐受。
(2)镰刀形红细胞贫血病

血液里的镰刀形红细胞
此病常认为是常染色体隐性遗传,更像不完全显性,致病基因的纯合体贫血严重,发育不良,关节、腹部和肌肉疼痛,多在幼年期死亡。但带有致病基因的杂合体大部分是无症状,或有的仅有轻度的贫血。但是,如果这种杂合体的人处于高原地区或长时间进行大强度运动训练而导致体内缺氧时,红细胞就会发生“镰变”,阻塞血管,引起全身发烧,肌肉酸痛,大量红细胞被脾脏吞噬,血红蛋白下降,机体运输O2和CO2的能力降低,造成机体红细胞破坏和缺氧的恶性循环。这种杂合体的人在麻醉、输血、体力消耗等特殊情况下,往往会出现死亡。
(3)体位性(直)蛋白尿
此病患者肾脏无器质性病变,但长时间性站立、行走、体力劳动,或进行大运动量训练时尿蛋白量增多,造成体质虚弱、无力。
(4)肝糖原贮积症
患儿在新生儿期就可能出现进行性肝肿大,喂养困难,生长发育迟缓,2岁内易出现严重低血糖症,并伴有惊厥。患儿身矮、肌肉松弛且发育不良,颊和臀部脂肪组织增多。有乳酸中毒,高脂血症,生长迟滞。肝活检查可见有大量糖原和脂质贮积。
提前预防低血糖症
(5)半乳糖血症
半乳糖血症是一种糖类代谢病。患儿出生后数日至数周即有呕吐、腹泻、黄疸,随之发生脱水、体重下降、肝损害,1~2个月后出现白内障。如不戒奶,则智力发育障碍,症状进行性加剧。正常婴儿体内的半乳糖经肠道吸收后,在肝内转变成1-磷酸葡萄糖而被利用。患儿因缺乏1-磷酸半乳糖尿苷转移酶,在进含乳食物后,血中1-磷酸半乳糖及半乳糖浓度明显升高。1-磷酸半乳糖在肝脏积聚可引起肝肿大,肝功受损;在脑的积聚可引起运动及智力障碍;血中半乳糖升高可使葡萄糖释出减少,出现低血糖症。另外,晶体内半乳糖增多,激活醛糖还原酶,产生半乳糖醇,引起白内障。
(6)丙酮酸激酶缺乏症
婴儿型多在新生儿期即出现症状,黄疸与贫血都比较严重,黄疸可发生在生后两天内,甚至需要换血。肝脾明显肿大,生长、发育受到障碍,重者常需多次输血才能维持生命。但随年龄增大,血红蛋白可以维持在低水平,不再需输血。化验可见红细胞较大,非球型。红细胞丙酮酸激酶活性降低,常降至正常值的30%左右。此病纯合子发病,杂合子不显症状,故患者的父母(携带者)丙酮酸激酶活性也轻度降低。成人型症状很轻,常被忽视。多于合并感染时才出现贫血。丙酮酸激酶是红细胞中的一种酶,其功能是在糖无氧酵解过程中起催化作用。如该酶缺乏可使红细胞内ATP的产生减少,从而影响红细胞的寿命,引起溶血性贫血。

较大红细胞
ATP空间结构
(7)黑蒙性痴呆
此病是中枢神经系统有类脂物质积累引起的进行性智力障碍和视力障碍的遗传性疾病。婴儿出生后4~8个月间出现症状,最初表现为呆钝、淡漠,以后坐时头不能抬直,肌肉无力,体重逐渐降低,而且已有的运动反射也消失了,视力明显减弱以至于失明。眼底检查黄斑部有樱桃红色点,周围有变性细胞构成的灰白色圈,视神经乳头苍白,几个月后可完全失明,痴呆和不能运动。随着病情的进展,肌肉出现强直痉挛,反射活跃。腱反射亢进、颈肌反射呈阳性,常常伴有惊厥。病儿有时狂笑,有时高叫。1岁时很胖,最后极度衰竭、消瘦,但肝脾不增大,无严重贫血。
(8)高雪氏病

患有角弓反张的婴儿
因脂肪代谢紊乱,造成网状内皮细胞有脑苷脂类沉积,所以此病可分为急性和慢性两种。急性型,也称婴儿型。在婴儿1岁内起病,肝脾肿大,贫血,发育迟缓,出现神经系统症状,如意识障碍、角弓反张、四肢强直、集合性斜视等,亦可发生惊厥。慢性型:也称幼儿型,起病较慢,可在任何年龄发病,以学龄前发病较多。起初出现肝肿大及轻度贫血,以后脾肿大,贫血加重,血小板及白细胞明显减少,也伴有皮肤、黏膜出血。长骨关节可有隐痛和局限性压痛,偶尔会发生病理性骨折,X线检查可见骨质疏松。此病患者葡萄糖神经酰胺酶缺乏,神经鞘脂类代谢障碍,葡萄糖脑苷脂蓄积在网状内皮系统细胞中,主要侵犯肝、脾、淋巴结及骨髓。
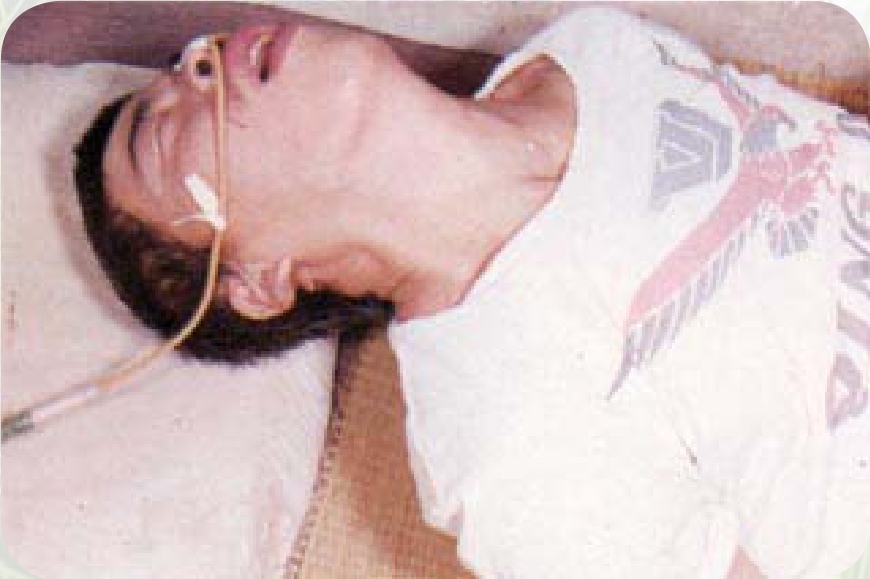
角弓反张症状
除此之外,常染色体隐性遗传病常见的还有着色性干皮症、家族性痉挛性下肢麻痹、先天性聋哑、侏儒症、呆小症、半乳糖血症、黑白内障、白痴、胃溃疡、先天性鱼鳞皮病、遗传性小头畸形、裂唇和裂腭、肥胖生殖无能综合征、粘多糖过多症、高度远视、高度近视、婴儿型青光眼等。知识小百科
“一个基因、一个酶”
一个基因一个酶这是一种学说,认为一个基因仅仅参与一个酶的生成,并决定该酶的特异性和影响表型。1941年,由G·W·Bea-dle和E·L·Tatum进行了链孢霉中生化反应遗传控制的研究;进而使应用各种生化突变型对基因作用的研究有了发展。1945年,Beadle总结了这些结果,提出了一个基因一个酶的假说。
基因链
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










