



第二节 遗传性酶病
遗传性酶病是指由于遗传性酶缺陷引起的,使机体的代谢过程不能正常进行,最终导致的疾病,也称先天性代谢病。至今已发现2 000多种遗传性酶病,其中200多种病的酶缺陷已清楚,其遗传方式多为常染色体隐性遗传,少数为常染色体显性遗传和X连锁隐性遗传。
一、遗传性酶病的发病机理
酶是具有催化作用的蛋白质,人类的遗传性状是基因通过合成特定的酶控制机体新陈代谢而形成的。基因突变可导致酶的蛋白质结构异常,基因调控系统突变可导致酶的合成数量减少,两者均可导致遗传性酶缺乏,引起代谢紊乱。仅少数酶的活性增高可导致遗传性酶病。
基因突变导致酶活性改变的可能原因如下。
(1)结构基因突变:①导致酶动力学特性改变,表现为酶与底物的亲和力降低,与抑制物亲和力增高;②导致酶稳定性降低,表现为酶降解速率加快。
(2)调节基因突变:导致酶合成速率减慢。
(3)影响翻译后修饰和加工。
二、常见的遗传性酶病
(一)苯丙酮尿症
苯丙酮尿症(phenylketonuria,PKU)是一种氨基酸代谢病,即氨基酸代谢过程中酶遗传性缺乏引起的氨基酸代谢缺陷,具体由苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)遗传性缺乏引起。本病是造成智力低下的常见原因之一,呈常染色体隐性遗传。该病在我国的发病率约为1/16 500。
在正常人体内,苯丙氨酸通过苯丙氨酸羟化酶转变为酪氨酸,继而生成黑色素(图8-5)。苯丙酮尿症患者是由于肝中苯丙氨酸羟化酶的基因突变导致肝细胞中苯丙氨酸羟化酶活性降低或完全丧失,若苯丙氨酸羟化酶的活性小于1/10,可阻断苯丙氨酸转化成酪氨酸,苯丙氨酸经旁路代谢产生苯丙酮酸、苯乳酸、苯乙酸等代谢产物,由尿液和汗液排出,使患儿体表、尿液有特殊的“鼠尿味”,产生经典型苯丙酮尿症。若苯丙氨酸羟化酶部分缺乏,将导致轻度苯丙酮尿症。旁路代谢产物累积可抑制L-谷氨酸脱羟酶的活性,使γ-氨基丁酸生成减少,同时还可抑制5-羟色氨脱羧酶的活性,影响5-羟色氨生成,从而影响大脑发育。
经典型苯丙酮尿症患儿出生时基本正常,3~4个月时,逐渐出现智力发育不全,未治愈者将发展为白痴。患儿步伐小,姿势似猿猴,肌张力亢进,易激动,甚至惊厥,多数有脑电图异常。90%以上的患者表现为毛发淡黄,皮肤白皙,甚至虹膜呈黄色(白种人呈蓝色)。此外,患儿的尿液和汗液中有一种特殊的鼠尿味。如能早期明确诊断,该病可采用低苯丙氨酸饮食等饮食治疗方法控制病情发展。
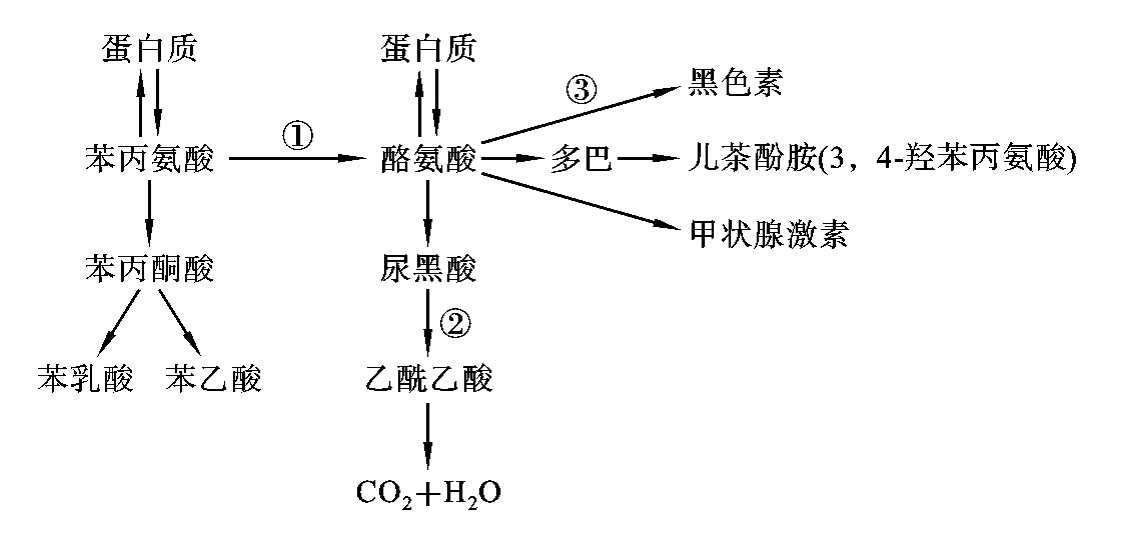
图8-5 苯丙氨酸和酪氨酸代谢
注:①苯丙氨酸羟化酶缺乏→苯丙酮尿症;②尿黑酸氧化酶缺乏→尿黑酸尿症;
③酪氨酸酶缺乏→白化病。
苯丙氨酸羟化酶基因定位于12q24.1,其cDNA全长90kb,有13个外显子和12个内含子。该基因主要在肝脏中表达。目前已发现200多种基因突变,其中多数为错义突变,其余为缺失突变、插入突变和移码突变。
(二)尿黑酸尿症
尿黑酸尿症(alkaptonuria)是一种较少见的先天性氨基酸代谢障碍疾病,尿黑酸氧化酶先天性缺乏导致尿中排出尿黑酸(2,5-二羟苯乙酸)。患者表现为黑色尿及脊柱和大关节的退行性关节炎。由于患者体内缺乏尿黑酸氧化酶,尿黑酸不能氧化成乙酰乙酸和延胡索酸,导致大量尿黑酸从尿中排出。刚排出的尿是无色的,与空气接触后,大量尿黑酸被氧化,尿液迅速变成黑色。新生儿期患者常在尿布上出现紫褐色斑点,日久渐变成黑褐色;儿童期患者除排出尿黑酸尿外,并无特殊症状;成人期患者除排出尿黑酸尿外,内源性尿黑酸自身氧化形成的产物沉淀于软骨、结缔组织和胶原组织,导致褐黄病,表现为皮肤、面颊、耳廓、巩膜等处有弥漫性色素沉着,上腭出现蓝色或黑色的色素斑块,若累及关节会使大关节和脊柱发生退行性改变。
本病为常染色体隐性遗传,发病率约为1/250 000。尿黑酸氧化酶基因定位于3q21—q23。
(三)眼皮肤白化病
眼皮肤白化病(oculocutaneous albinism,OCA)是指先天性皮肤、毛发色素缺乏的疾病,患病率为1/20 000~1/10 000。
患者表现为皮肤呈乳白色,毛发为白色、银白色或淡黄色,在黑人患者中皮肤可出现黑色痣。患者瞳孔淡红,虹膜呈淡红或浅黑色,视网膜无色素,畏光,眼球震颤,严重视力低下者不能由佩戴眼镜加以矫正。
眼皮肤白化病具有遗传异质性。目前已确定的4型眼皮肤白化病(OCA1~OCA4)均呈常染色体隐性遗传。患者因缺乏酪氨酸酶,不能正常形成黑色素而导致皮肤、毛发白化。黑色素能阻挡阳光中的紫外线,使机体内部器官免受伤害,但白化病患者因缺乏黑色素,不能经受长时间日光照射,否则易产生皮肤癌。现已知致病基因(酪氨酸酶基因)定位于11q14-q21,OCA1和OCA2是OCA最常见的两种类型。迄今已确定了至少142种导致OCA1的致病性突变。OCA2也称阳性眼皮肤白化病,是最常见的白化病类型,是由定位于15q11.2-q12的P蛋白基因突变所致。
某些眼皮肤白化病患者的毛发和皮肤均正常,只有眼色素缺乏,遗传方式为X连锁隐性遗传,基因定位于Xp22.3。
(四)半乳糖血症
半乳糖血症(galactosemia)是指由于遗传性酶缺乏引起的糖代谢病,现已发现了Ⅰ型、Ⅱ型和Ⅲ型三种类型,Ⅰ型也称经典型。
半乳糖血症Ⅰ型由半乳糖-1-磷酸尿苷转移酶遗传性缺乏引起。新生儿患病率为1/6 000~1/4 000。由于此酶缺乏,半乳糖-1-磷酸及半乳糖在脑部积累而引起智力障碍,在肝脏积累而引起肝硬化,在血中积累可使葡萄糖释出量减少,出现低血糖症。半乳糖以晶状体形式积累,在醛糖还原酶的作用下产生半乳糖醇,导致晶状体代谢障碍,形成白内障。患儿出生后用乳类喂养数日,即出现呕吐、腹泻,表现为对乳类不耐受,一周后逐渐出现肝大、黄疸、腹腔积液和白内障,数月后出现明显智力发育不全症状,大多数患儿于新生儿期因感染死亡。该病呈常染色体隐性遗传,半乳糖-1-磷酸尿苷转移酶基因定位于9q13。
半乳糖血症Ⅱ型由半乳糖激酶缺乏引起,其症状较半乳糖血症Ⅰ型的轻,主要表现为青年型白内障,血中半乳糖含量增高,但无肝及脑损害,尿中可出现半乳糖和半乳糖醇,但无氨基酸和蛋白质。临床表现变化不一,有的患儿肝、脾肿大,无黄疸,有的黄疸明显,智力发育正常或迟缓。半乳糖激酶基因定位于17q24。
半乳糖血症Ⅲ型由半乳糖尿苷2-磷酸-4-表异构酶缺乏引起,该酶基因定位于1p36—p35。临床表现不一,可无临床症状或症状与半乳糖血症Ⅰ型的类似。半乳糖血症Ⅱ型、Ⅲ型均为常染色体隐性遗传,它们的患病率较半乳糖血症Ⅰ型的低。
(五)糖原贮积症
糖原贮积症(glycogen storage disease,GSD)是指由于糖原分解过程中的酶缺乏引起的疾病。糖原又称肝糖、动物淀粉,是由许多葡萄糖结合而成的带支链的大分子多糖,主要存在于肝脏和肌肉中。糖原的分解过程涉及多种酶,是复杂的酶促反应,其中任何一种酶的缺乏均可致病。目前已发现13种类型的糖原贮积症(表8-3),以糖原贮积症Ⅰ型(von Gierke病)最为常见。
表8-3 糖原贮积症的类型
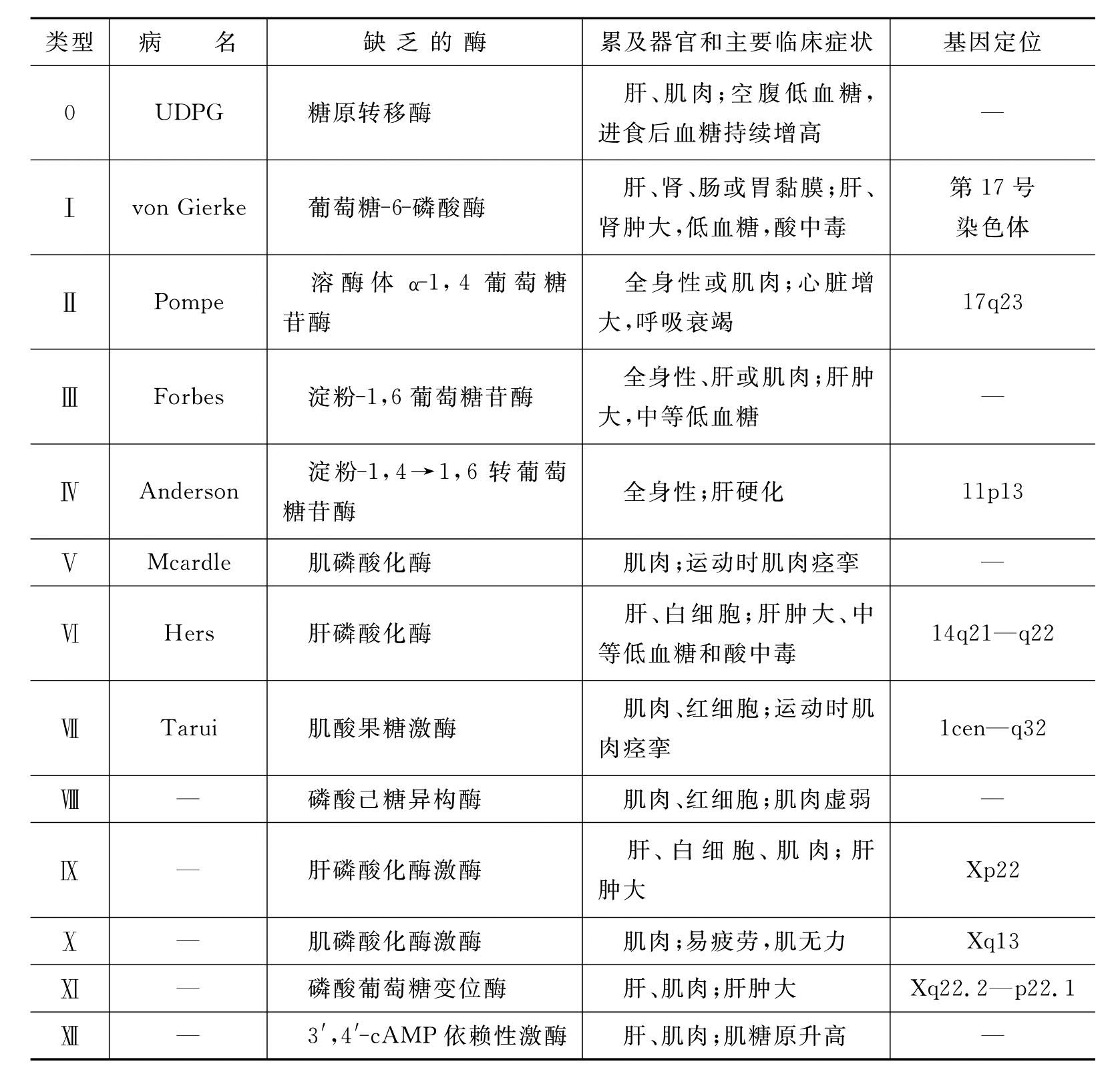
糖原贮积症Ⅰ型,又称肝肾型糖原贮积症,1929年由von Gierke首次报告。本病是由于肝、肾、肠组织完全缺乏葡萄糖-6-磷酸(G-6-P)酶缺乏引起的,患者的病变主要累及肝和肾,不侵犯骨骼肌和心脏。
葡萄糖-6-磷酸酶缺乏使葡萄糖-6-磷酸不能转变成葡萄糖,糖原分解代谢受阻,却通过可逆反应合成过多的肝糖原。葡萄糖-6-磷酸另外通过酵解途径,产生大量丙酮酸和乳酸,导致酸中毒。肝糖原在肝细胞中聚集,导致患儿易怒、脸色苍白、发绀、喂养困难及低血糖抽搐、肝大、发育迟缓等。患儿在5~6岁后以出血、感染为主要症状。该病为常染色体隐性遗传,葡萄糖-6-磷酸酶的基因定位于第17号染色体。
(六)黏多糖贮积症
黏多糖贮积症(mucopolysaccharidosis,MPS)是一种溶酶体贮积病,溶酶体贮积病包含黏多糖贮积症、鞘磷脂贮积症、糖脂质贮积症等多种类型。
黏多糖是由蛋白质和氨基多糖构成的糖蛋白,因氨基多糖含有较多的糖醛酸和硫酸基团,所以呈酸性。一条蛋白质肽链上可同时存在几种不同的氨基多糖链,还可进一步聚合成更大的分子,结构十分复杂。
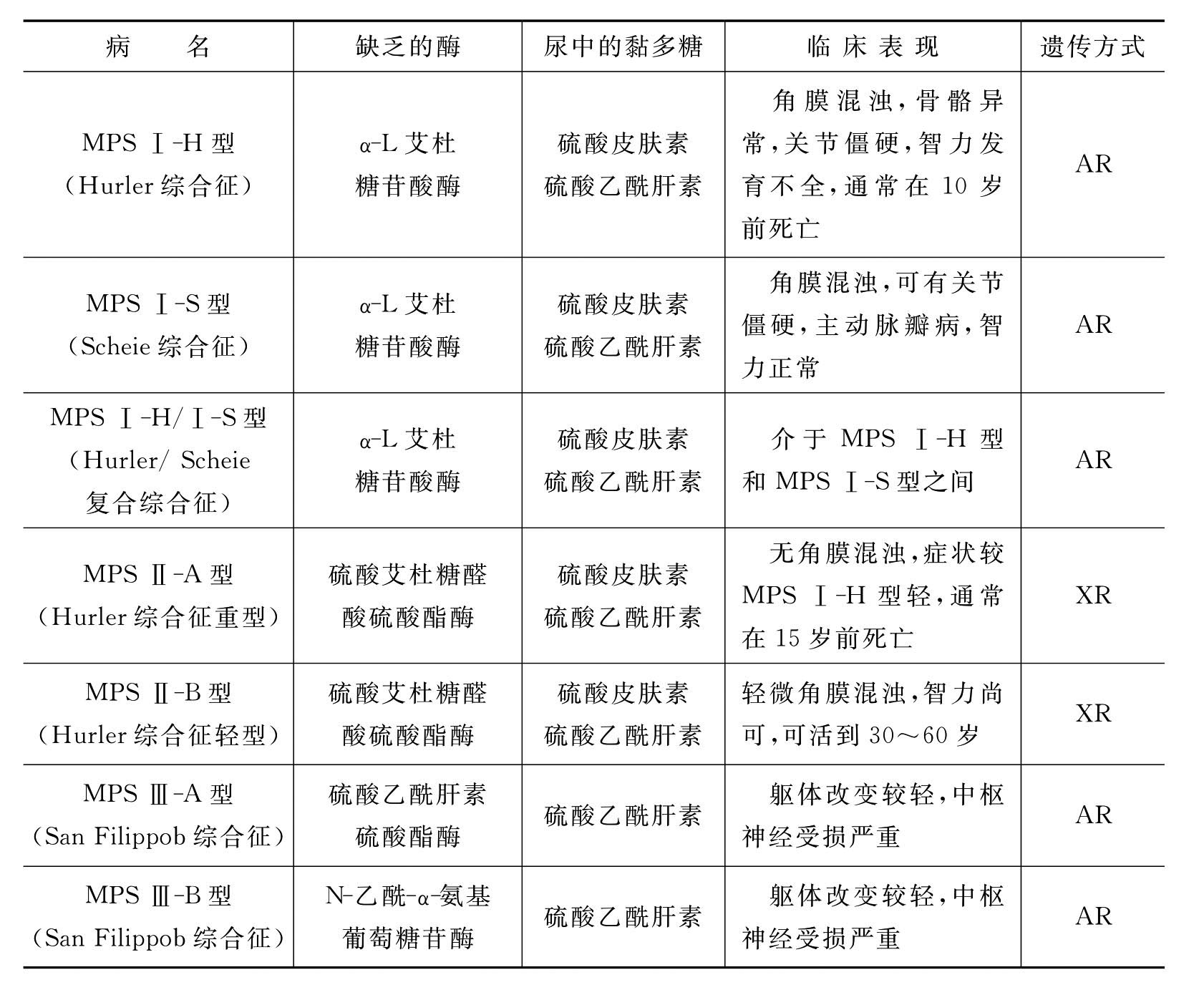
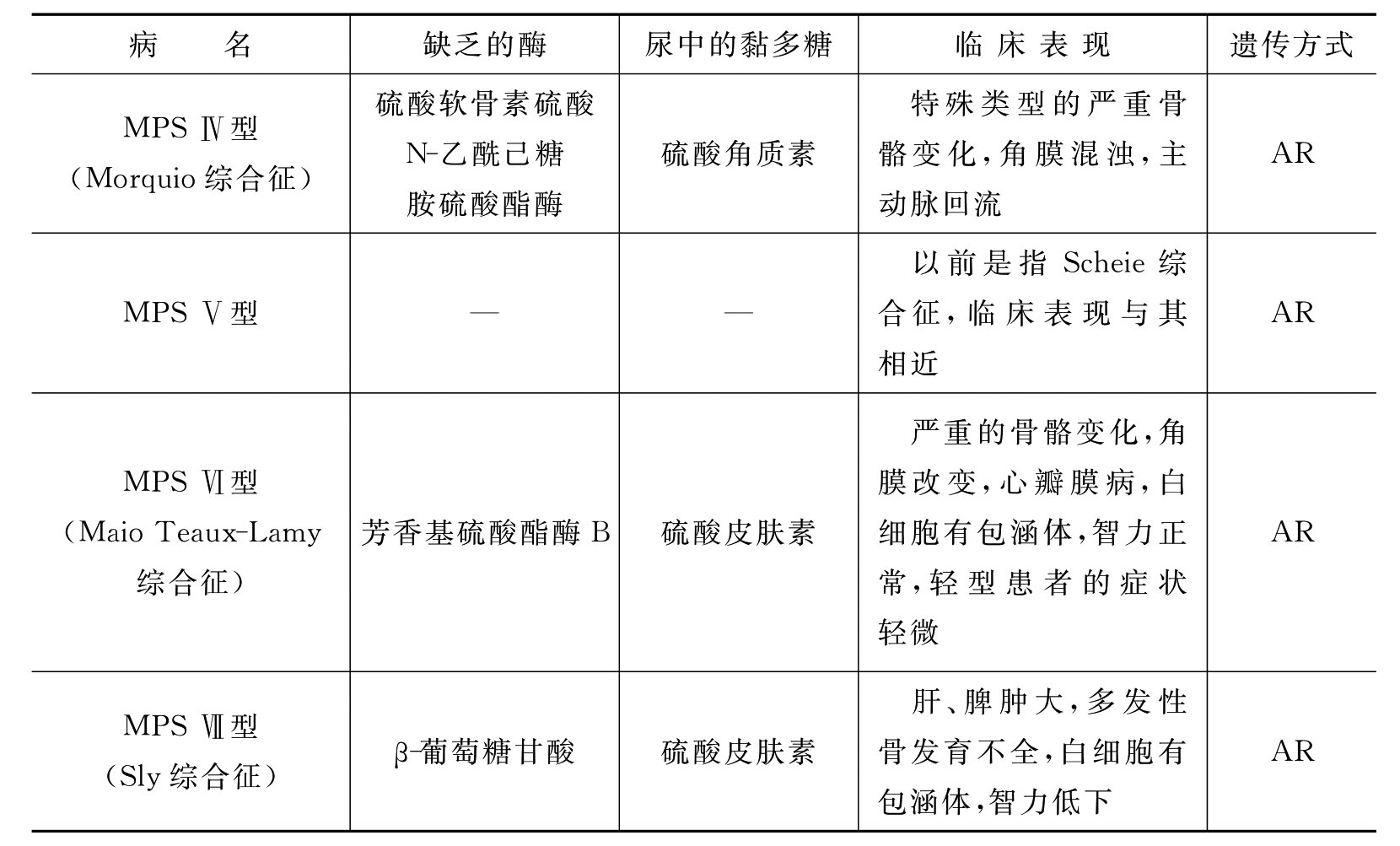
MPS是由于特定的糖苷酶或硫酸酯酶遗传性缺乏,导致酸性黏多糖的部分分解产物在机体各组织中贮积而致病的。患者面容粗陋,骨骼畸形,甚至伴有智力障碍和肝、脾、心等器官的损害。贮积的酸性黏多糖大多是由硫酸皮肤素(DS)产生的,是结缔组织的成分,DS主要分布于皮肤、韧带、动脉及心瓣膜。黏多糖贮积症根据缺乏的酶的种类不同,可分为下列几种类型(表8-4)。
表8-4 黏多糖贮积症分型及主要临床表现
续表
(七)泰-萨氏病
泰-萨氏病(Tay-Sachs disease)又称家族性黑矇性白痴、GM2神经节苷脂贮积症,是由于氨基己糖苷酶A缺乏,使GM2神经节苷脂分解成GM3神经节苷脂和N-乙酰氨基半乳糖代谢受阻,导致GM2神经节苷脂累积所致。患者初起症状为听觉过敏,早期可见视网膜黄斑变性,视网膜有樱桃红斑点,进行性失明,常有局部性或全身性抽搐及痴呆。患儿表现为进行性肌张力减退、衰弱、生长迟缓,晚期完全瘫痪,出现恶病质,平均存活25.9个月。
该病为常染色体隐性遗传。已知氨基己糖苷酶A基因定位于15q23—q24。已检出碱基替换、缺失和移码突变是氨基己糖苷酶A的基因突变类型。
(八)戈谢病
戈谢病是由于葡萄糖脑苷脂酶缺乏导致葡萄糖脑苷脂贮积于网状内皮系统细胞中引起的,主要损伤肝、脾、淋巴结及骨髓。
本病可分为急性婴儿型和慢性型两种类型。急性婴儿型患者于1岁内发病,主要表现为肝和脾肿大、贫血、发育迟缓、全身性肌张力过度、角弓反张、四肢强直、意识障碍、集合性斜视及吞咽困难、呕吐、喉头痉挛和呼吸困难,也可发生惊厥等,通常2岁前死亡。慢性型多于学龄前发病,主要表现为肝和脾肿大、贫血、骨骼系统受损,但不累及神经系统。患者可存活十几年至数十年。
本病呈常染色体隐性遗传,已知葡萄糖脑苷脂酶基因定位于1q21。羊水细胞测定酶活性或检测分子变异可用于该病的产前诊断。
(九)自毁容貌综合征
自毁容貌综合征也称Lesch-Nyhan综合征,患者遗传性缺乏次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HPRT),使5-磷酸核糖-1-焦磷酸上的磷酸核糖基不能正常地转移到鸟嘌呤和次黄嘌呤上,导致鸟嘌呤核苷酸和次黄嘌呤核苷酸(肌苷酸)生成受阻,不能有效反馈抑制嘌呤前体5-磷酸核糖-1-胺的生成,致使嘌呤合成加快、尿酸增高、代谢紊乱而致病。
患者的临床表现常有高尿酸症、血尿、痛风症状等,伴有智力发育不全、舞蹈样动作和强迫性自身毁伤行为,因此本病也称自残综合征。患者大多在儿童时期死于感染和肾衰竭,一般活不过20岁。若HPRT部分缺乏,可引起高尿酸血症和痛风,不出现以上症状。
本病为X连锁隐性遗传,发病率约为1/38 000。HPRT基因定位于Xq26—q27.2,已发现50多种突变。本病可在DNA水平上进行产前诊断。
小结
分子病是由于遗传缺陷造成的蛋白质结构或数量异常所引起的疾病,此种异常有的可以向后代传递。分子病的种类较多,主要包括血红蛋白病、血友病、假肥大型肌营养不良和家族性高胆固醇血症等。其中血红蛋白病是严重危害人类健康的一种分子病,据统计,全世界约有1.5%的人带有血红蛋白病的致病基因,每年出生的各类重型患儿多达20万,本病也是研究的最深入、最透彻的分子病,目前已能应用遗传工程的方法作出血红蛋白病等分子病的产前诊断。血友病是一种遗传性的因凝血因子缺乏形成的出血性疾病,不同类型的临床表现轻重也不一样,近年来我国在应用基因疗法治疗血友病方面已取得突破性的进展。
遗传性酶病也称先天性代谢病,是指由于遗传性酶的缺乏引起机体代谢紊乱而导致的疾病。根据代谢物的生化性质,可分为氨基酸代谢病、糖代谢病、脂类代谢病、核酸代谢病等。苯丙酮尿症、尿黑酸尿症、眼皮肤白化病属于氨基酸代谢病;半乳糖血症、糖原贮积症、黏多糖贮积症属于糖代谢病;泰-萨氏病、戈谢病属于脂类代谢病;自毁容貌综合征属于核酸代谢病。
每种病均有特定的临床症状和遗传方式,但都是由于缺乏某一种酶基因引起,致使体内的某种生化反应不能正常进行,引起代谢紊乱。大多缺乏的酶基因均已定位,这为临床治疗和研究提供了理论依据。
能力检测
一、选择题
1.血红蛋白分子是由( )构成的球形四聚体。
A.一对类α链和一对类β链 B.两对类α链
C.两对类β链 D.一对珠蛋白链
2.正常人二倍体细胞中共有( )个α基因。
A.1 B.2 C.3 D.4
3.Hb Catonsville是由于α珠蛋白基因第37和第38密码子之间插入了一个谷氨酸的密码子形成的,这种基因突变属于( )。
A.单个碱基置换 B.移码突变
C.整码突变 D.不等交换
4.在α地中海贫血中,(-/-α)代表的是( )。
A.4个α基因全部缺失 B.3个α基因缺失
C.2个α基因缺失 D.1个α基因缺失
5.由于基因突变,影响了内含子的正确位点剪接,产生异常的mRNA,所发生的突变是( )。
A.内含子突变 B.外显子突变
C.启动子突变 D.加工修饰点突变
6.血友病是由于哪一种凝血因子缺乏引起的?( )
A.凝血因子Ⅸ B.凝血因子Ⅺ
C.凝血因子Ⅳ D.凝血因子Ⅷ
7.苯丙酮尿症是由于缺乏( )引起的。
A.苯丙氨酸羟化酶 B.尿黑酸氧化酶
C.酪氨酸酶 D.L-谷氨酸脱羧酶
8.尿黑酸尿症是由于缺乏( )引起的。
A.苯丙氨酸羟化酶 B.尿黑酸氧化酶
C.酪氨酸酶 D.L-谷氨酸脱羧酶
9.眼皮肤白化病是由于缺乏( )引起的。
A.苯丙氨酸羟化酶 B.尿黑酸氧化酶
C.酪氨酸酶 D.L-谷氨酸脱羧酶
10.半乳糖血症是由于缺乏( )引起的。
A.半乳糖激酶
B.半乳糖-1-磷酸尿苷转移酶
C.半乳糖尿苷-2-磷酸-4-表异构酶
D.焦磷酸酶
11.自毁容貌综合征是一种( )代谢病。
A.氨基酸 B.糖 C.脂类 D.核酸
二、名词解释
1.分子病
2.血红蛋白病
3.异常血红蛋白病
4.受体
5.受体蛋白病
6.不等交换
7.遗传性酶病
三、简答题
1.异常血红蛋白病有哪些类型?
2.异常血红蛋白病产生的基因突变有哪些?
3.α地中海贫血有哪几种类型?各型有哪些主要临床表现?
4.导致β地中海贫血的基因突变有哪几种?
5.由低密度脂蛋白(LDL)受体基因突变引起的低密度脂蛋白受体缺陷有哪些表现?
6.基因突变引起酶活性改变的可能原因有哪些?
(李晓光)
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。











