



第一节 遗传性肾小球病
遗传性肾小球病(hereditary glomerular nephropathy),也称原发性遗传性肾小球病。在肾活检病例中,可见到遗传性肾炎(Alport综合征)、薄基膜肾病、甲髌综合征、先天性肾病综合征、Ⅲ型胶原肾小球病和纤连蛋白肾病等。根据我组对资料完整的7 908例肾活检病例的统计,以薄基膜肾病为最常见,其次是遗传性肾炎,而其他类型的遗传性肾小球病均十分少见。
一、遗传性肾炎(Alport综合征)
遗传性肾炎(hereditary nephritis),又称Alport综合征(Alport syndrome)。本病于1902年首先由Guthrie报道,作者发现在同一个家族中,有12个成员先后发生持续性血尿,并最终发展为肾衰竭。1923年,Herst又对其作了跟踪报道,并命名其为“遗传性家族性先天性出血性肾炎”(hereditary familial congenital hemorrhagic nephritis)。至1927年,由Alport等再对其作报道,不仅肯定其特征性表现为进行性肾炎,还发现患者和其他家族成员中,合并存在耳聋病例和男性患者的病情明显重于女性的特点,并确认其是一种X染色体连锁显性遗传的疾病。此后,这一疾病先后在世界各地被陆续报道。目前,对这一疾病的名称尚不统一,部分学者将伴有耳聋表现的肾炎称为Alport综合征,而对于不伴耳聋的病例,则命名其为遗传性肾炎。然而,目前多数学者已不再对其进行严格区分而将这两种名称通用,故本书也将其合在一起作介绍。在我组收集的7 908例肾活检病例中,确诊为遗传性肾炎者6例,仅占0.08%。
遗传性肾炎最常见的临床表现是无症状性血尿,多数患者可自5岁起出现间隙性血尿,偶尔其症状也可发生于婴儿期,部分病例可伴有发作性肉眼血尿和进行性蛋白尿,偶尔也可呈现脓尿。实验室检查可发现在部分患者血清中,存在抗基膜Ⅳ型胶原(ColⅣ)NC1,即Goodpasture抗原的抗体。本病在男女性别中的发病概率均等,但女性患者的病情明显轻于男性,甚少发展成慢性肾衰竭,而男性患者,如不进行有效的透析治疗或接受良好的移植肾,则常在40岁之前死于尿毒症。偶尔患者也可合并发生眼科疾患,如双侧前圆锥形晶状体、白内障和晶状体膜破裂等。
【病因和发病机制】
本病是一种与异源性遗传基因密切相关的疾病。研究表明,其大多数(80%~85%)患者的遗传方式为X染色体连锁显性遗传,但也有部分患者可能与ColⅣα链生成相关基因所在染色体,如编码α3、α4链的基因Col 4A3、Col 4A4所在的2q35改变有关,可为常染色体显性遗传,或极少数呈常染色体隐性遗传性疾病。本病的发病机制仍不明,但近年来随着病理学、生物化学和分子遗传学等研究的进展,已有足够的证据表明,其发病可能与一些调控基膜(如GBM、晶状体膜和内耳组织等)主要成分ColⅣ合成和修复,尤其是位于Xq22位点的编码ColⅣα5链的结构基因(Col 4A5)发生突变或缺失所致,进而可引起单聚体或双聚体的ColⅣC末端NC1功能区(包含Goodpasture抗原)的缺损或其结构改变,从而造成其与ColⅣ交联的不足或不完整而致病,有学者确认其是导致患者GBM致密层呈现多层或分裂等形态改变及患者出现持续性血尿的重要机制之一。
【病理改变】
遗传性肾炎的早期,肾小球形态结构可为正常,或仅表现GBM弥漫性变厚,或伴有较多发育不成熟肾小球,肾小管可呈现灶性萎缩,肾间质灶性纤维化伴单个核细胞浸润。随着疾病的进展,GBM可明显增厚,可伴有血管襻节段性系膜细胞增生、节段性硬化(图6-1)伴透明变性,也可伴有新月体形成、血管襻与球囊壁发生粘连、部分肾小球硬化等病变,但其病灶多呈局灶性分布。肾间质内常可有灶性分布的泡沫细胞浸润(图6-2,6-3),一些作者认为其对本病诊断有一定参考价值,但不具特异性。肾小动脉内膜可显示轻度增厚等。

▲图6-1 遗传性肾炎(PAS ×200)
肾小球血管襻系膜基质增多,与球囊壁粘连伴纤维化,肾小管萎缩、基膜增厚及肾间质纤维化

▲图6-2 遗传性肾炎(HE ×200)
肾间质内泡沫细胞形成,伴灶性淋巴细胞浸润

▲图6-3 遗传性肾炎(EM ×1 500)
肾间质内呈现大量泡沫细胞浸润
免疫荧光检查显示,大多数病例的肾组织无免疫球蛋白、补体的沉积,偶有非特异性IgM和(或)补体的沉积。近年来国内外一些学者,应用免疫组化法检测肾小球和肾小管基膜ColⅣ的α1~α6链,发现患者GBM的α3~α5链的表达呈阴性,并认为其对本病作出正确诊断有极其重要的参考价值。
电镜检查在其典型病例中,常显示GBM节段性增厚(800~1 200 nm),其致密层出现分裂(splitting)或呈分层状(lamination)(图6-4),或表现为弥漫性变薄,可伴有裂隙(gaps)或裂口(breaks)(图6-5)形成,或GBM呈现形态不规则、厚薄不均(图6-6)或呈交叉状排列,其形态犹如“篮网状”(basket-weave pattern),其间可出现透明间隙(clear space),有时也可含直径约90 nm、呈黑色(即嗜锇性)的致密颗粒(可能为脂质)。有作者研究发现,其病变的严重性常与患者尿蛋白程度及临床经过呈密切相关性。部分病例在疾病早期,因致密层明显减少而致GBM变得菲薄,可仅为正常基膜厚的1/3,成为Alport综合征唯一的超微结构改变,但对本病诊断不具有特异性。只有当发现GBM显著增厚,伴分裂或呈分层状改变,而又可除外其他肾小球疾病时,才可考虑Alport综合征的可能。肾小管基膜也常可变厚,呈现分裂或分层状改变。

▲图6-4 遗传性肾炎(EM ×12 000)
肾小球血管壁基膜(BM)局部增厚,致密层呈分层状

▲图6-5 遗传性肾炎(EM ×18 000)
肾小球血管壁基膜厚薄不均,有一裂口(↑)形成

▲图6-6 遗传性肾炎(EM ×5 250)
示肾小球血管壁基膜形态不规则,厚薄不均
【鉴别诊断】
须与遗传性肾炎作鉴别的肾病,是一些可引起儿童期血尿的疾病,包括良性家族性血尿(薄基膜肾病)、散发性良性血尿等。典型的遗传性肾炎病例,只要有明确的家族史,并经肾活检组织的GBM ColⅣ的免疫组化和电镜检查,其诊断一般不难作出。然而对其一些早期病例,若在电镜检查中仅发现GBM变薄,则可能与这些疾病难以作出鉴别。近年来有文献报道,经采用抗Goodpasture抗原的单克隆抗体,或使用Goodpasture综合征患者的血清,对这些病例的肾组织做免疫荧光检查,发现其GBM和TBM呈线状分布的免疫荧光较正常肾组织明显减弱或呈阴性,或应用免疫组化法检测GBM的α1~α6(Ⅳ)链,当发现其GBM和TBM的α3~α5链表达呈阴性时,均有助于对遗传性肾炎的诊断。
【预后】
本病的预后主要取决于患者的性别和遗传方式。据文献报道,最终发生终末期肾衰竭的男性要比女性提早20~25年;而X染色体连锁显性遗传者的预后较常染色体显性遗传者要差。此外,其预后与是否合并耳聋有一定关系,合并耳聋的男性患者的预后比无耳聋者要差,然而在女性患者,其结果恰好相反。
二、薄基膜肾病
薄基膜肾病(thin basement membrane nephropathy),又称薄基膜综合征(thin basement membrane syndrome),即临床上所称的良性家族性血尿(benign familial hematuria)。本病于1966年,由McConville等首先以“家族性血尿”的名称进行报道。1973年,Roger等又报道了在同一家族中有4代8人,均以血尿为初发症状,其中有5人经肾穿活组织检查,证实其肾小球以GBM变薄(平均厚约150nm)为主要超微结构改变,并确认其是一种良性遗传性疾病。近年来,一些学者就其薄GBM的诊断标准(即平均厚度)进行了许多报道。在各组报道中,对正常人GBM厚度的检测结果有较大差异,如以Lang等报道的结果为最低(224~333 nm),而以Dische等报道的结果为最高(354~464 nm),这可能与他们各自制订的检测方法不同有关。近年来一些作者研究认为,GBM的平均厚度可自婴幼儿者约100 nm,至5岁以上者的300 nm,故确定正常人GBM的平均厚度>300 nm。因此,目前许多作者,将薄基膜肾病的诊断标准确定为GBM的平均厚度<250nm,但也有学者将其标准定为<300nm。根据国外文献制订的标准,我组对238例薄基膜肾病的GBM进行了检测,结果显示其GBM厚度均<250 nm。因此,笔者也将成人患者的GBM厚度确定为<250 nm,而对儿童患者的标准确定为<200 nm。这里值得一提的是,对于GBM厚度的测量方法必须严格掌握,作者的做法是在电镜下,任意选取5个以上的肾小球毛细血管襻进行摄片,选择足突与基膜呈垂直方向的基膜为测量点,每例的测量点为50个以上,最终求出其平均值。据上述标准,在我组资料完整的7 908例肾活检病例中,薄基膜肾病共有238例,占其总数的3.0%。
薄基膜肾病多见于青少年,但也可发生在成年人,男女性的发病概率均等。患者以持续性镜下血尿为主要表现,偶有肉眼血尿发作,也可伴有轻度蛋白尿,很少发生高血压和肾功能不全,也甚少进展到慢性肾衰竭。
【病因和发病机制】
薄基膜肾病的病因和发病机制均不明。据文献报道,多数家族性薄基膜肾病可能是在父子间进行传递的常染色体显性遗传性疾病。近年研究表明,肾GBM ColⅣ分布并无异常。也有少数患者,可能是常染色体隐性遗传(如Col 4A4基因突变)的Alport综合征的杂合子;或可能为不伴耳聋的进行性血尿性肾炎(progressive hematuric nephritis)或是经典性Alport综合征患者,常是ColⅣ基因突变所致;部分儿童或女性散发性血尿病例,可能是Alport综合征的早期表现。
【病理改变】
薄基膜肾病的肾小球形态结构基本正常,或伴有轻微的系膜增生。肾球囊腔和肾小管腔内有红细胞。免疫荧光检查常为阴性,有时可见散在颗粒状C3沉积。有作者报道,采用针对GBM ColⅣα链抗体作免疫荧光研究,发现其呈正常线状分布,在3例女性患者中,有2例为Alport综合征者,其标记物的荧光呈不连续性。电镜检查示GBM广泛而又规则的变薄(图6-6),偶见裂隙或裂口,但也有部分病例,在正常厚度基膜中,出现节段性变薄(图6-7)。足突结构常完好,多呈细长形(图6-8),部分病例的足突也可消失。

▲图6-7 薄基膜肾病(EM ×15 000)
肾小球血管壁基膜均匀变薄,呈节段性分层状
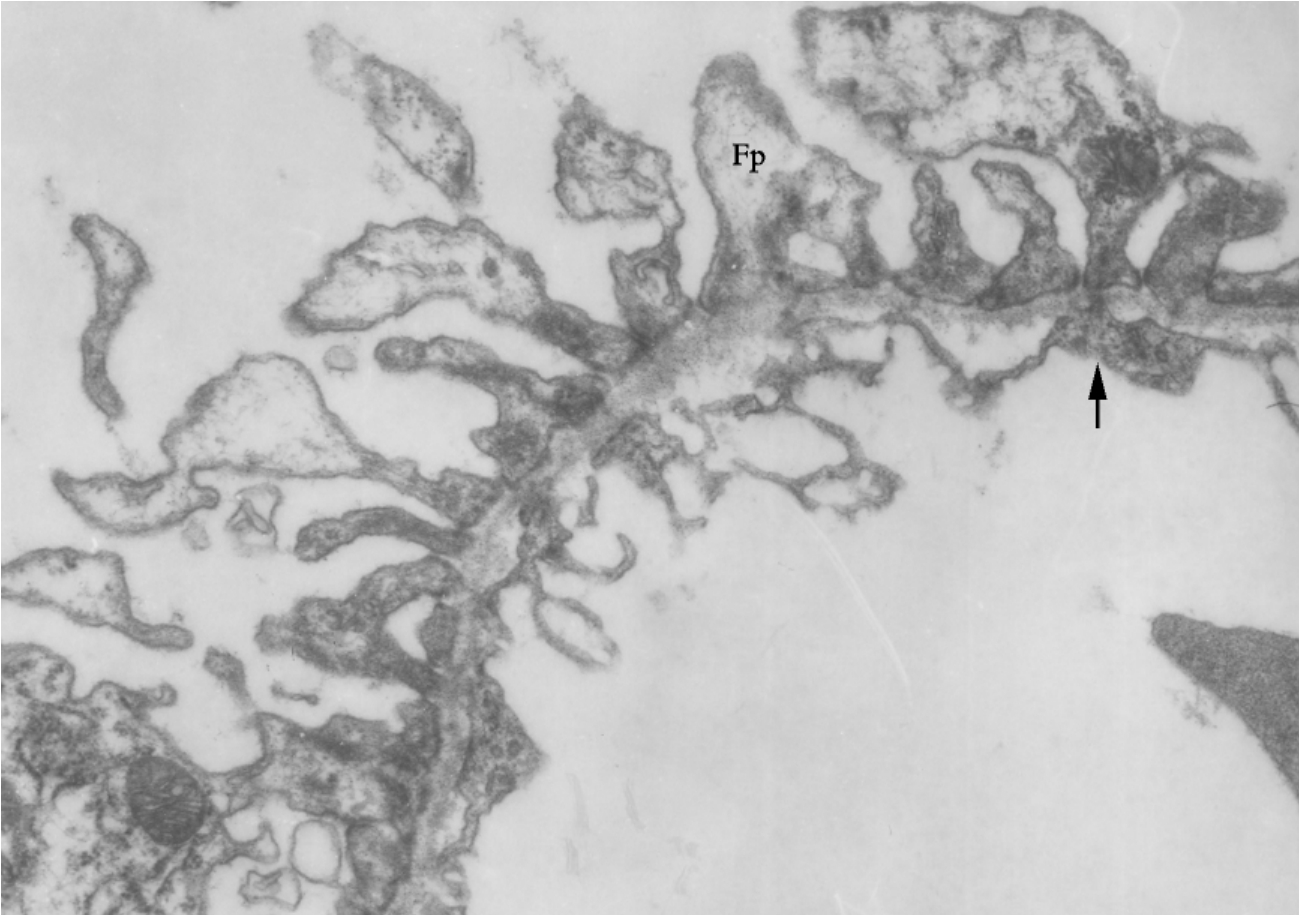
▲图6-8 薄基膜肾病(EM ×18 000)
肾小球血管壁基膜略有厚薄不均,有一裂口形成,足突(Fp)形态细长
【鉴别诊断】
正如前述,GBM广泛变薄也可见于Alport综合征病例的早期,然而后者常伴有Goodpasture抗原表达减弱或缺如、基膜分裂或分层明显。节段性GBM变薄也常可引起肾小球毛细血管腔扩张的其他肾小球病,如IgA肾病、糖尿病肾病等,然而其分布不呈广泛性,且伴有其他肾小球病的各种形态改变。
【预后】
薄基膜肾病在家族性血尿病例中,其长期预后大多良好,而对某些散发病例的预后尚不明,然而据国外学者对一组14个病例的报道,也有4例出现肾功能损害。
三、甲-髌综合征
甲-髌综合征(nail-patella syndrome,NPS),又称遗传性骨软骨发育不良(hereditary osteochondrosis),是一种十分少见的遗传性疾病。NPS多发生于婴幼儿,临床表现以指(趾)甲、骨骼发育不良或不发育为主要特征,常见者有指甲(尤其是拇指或拇趾)、髌骨、桡骨头发育不良和髂骨角形成伴膝关节、肘关节畸形等。肾脏累及常在疾病发生的多年之后,其发生率为30%~40%,主要表现为血尿、蛋白尿,也可致肾病综合征。
【病因和发病机制】
1955年,NPS首先被确定为一种与ABO血型基因连锁的常染色体显性遗传性疾病。以后又确定其与腺苷酸激酶(adenylate kinase,AR 1)基因位点连锁,并定位于9q34。近年来,又有学者确认其原发性缺陷发生在结缔组织,并伴有GBM的改变,并认为其可能为循环胶原前身物在肾小球内沉陷所致。故目前有作者,将位于9q34的TGF-β1受体基因和前B细胞白血病转录因子3基因(pre-B cell leukemia transcription factor 3 gene,PBX3),视为与NPS发病相关的候选基因,因为已知TGF-β与调控胶原合成有关,而PBX3则广泛表达于胚胎和成熟组织,可能其与组织定型有关。
【病理改变】
本病初期,肾小球的形态结构可为正常。随病程的延长,肾小球可出现轻微病变,如系膜基质增多、GBM不规则增厚,乃至发生肾小球局灶节段性硬化或球性硬化,伴肾小管萎缩、肾间质纤维化和肾细小动脉非特异性改变。免疫荧光检查结果常为阴性,或仅显示局灶节段性分布IgM和C3沉积。电镜检查显示GBM增厚,伴有不规则的电子透明区,可呈“虫蛀”状(moth eaten appearance)改变,内含深色纤维状物,伴有周期性,也可分布于肾小球系膜区或上皮下、内皮下区。
【鉴别诊断】
肾小球胶原纤维局限性沉积,可发生于各种不同类型肾炎的局灶节段性硬化或瘢痕区,这与NPS所出现的肾小球内,尤其是GBM内广泛沉积明显不同;而且这也是与不伴有甲-髌发育不全表现的Ⅲ型胶原肾小球病进行鉴别的重要超微结构标志之一。
【预后】
NPS的预后,取决于肾小球病变的严重性。在一些家族中,若伴发持续性、中度蛋白尿,甚至出现肾病综合征者,或据报道伴有膜性肾炎、IgA肾病、Goodpasture综合征和坏死性血管炎时,其预后则较差。据报道有1/4~1/3患者,至20~40岁时可进展至终末期肾。
四、弥漫性系膜硬化症
弥漫性系膜硬化症(diffuse mesangial sclerosis,DMS),又称法国型先天性肾病综合征(congenital nephrotic syndrome of French type)。于1973年,首先由Habib等在法国首先报道,此后被世界各国学者所证实。在我组收集的万余例肾活检病例中,只见1例。本病是一种呈家族分布、由常染色体隐性遗传的肾小球病。目前其发病机制仍不明,鉴于其可合并于肾母细胞瘤、假两性畸形的Drash综合征病例,且当今已肯定位于13p13染色体的WT1基因突变与患儿肾母细胞瘤的发生有关,因此有作者推测,本病的发生可能与WT1基因等改变有一定相关性。
婴儿发病多表现为肾病综合征,一般在出生3个月后发病。部分(近1/3)患者可合并肾外表现,如眼球震颤、神经发育迟缓、白内障、小头畸形、近视和心律失常等。肾活检组织最突出的组织病理学改变,是肾小球系膜基质增多,甚少伴系膜细胞增生。随病情发展,病变肾小球的毛细血管襻演变成呈实心的团块(图6-9),其PAS染色呈强阳性。肾小管可发生灶性萎缩,伴肾间质纤维化及炎症细胞浸润。免疫荧光检查结果为阴性。电镜检查显示,肾小球系膜基质明显增多,可伴有GBM不规则增厚。本病预后较差,如不进行肾移植,患儿多在3岁前因感染或肾衰竭而死亡。

▲图6-9 弥漫性系膜硬化症(HE ×100)
肾小球血管襻呈均质实心团块状,血管腔狭窄或闭塞,部分肾小管管腔扩张,伴少量蛋白、颗粒管型
五、Ⅲ型胶原肾小球病
Ⅲ型胶原肾小球病(type Ⅲ collagen glomerulopathy)或胶原Ⅲ型肾小球病(collagen typeⅢglomerulopathy),又称胶原纤维性肾小球病(collagenofibrotic glomerulopathy)。于1979年,本病首先由日本学者报道,此后在世界各地均有发现。据文献的不完全统计,其在全世界的病例数已有30余例,国内报道(包括我组1例)3例。本病主要的病理特点是胶原纤维在肾小球内的广泛沉积。因其病变与甲-髌综合征相似,故最初曾有作者认为,是甲-髌综合征的一种亚型或变异型。然而,目前多数作者根据其临床及遗传学的特点,已确定其为一种新的肾小球病。本病的发病机制尚不明确,但多数作者根据部分病例有明确的家族史和体内H因子缺失,认为其是一种常染色体隐性遗传性疾病。
患者可为婴儿或成年人,男女均可累及。临床表现多为持续性蛋白尿,可致肾病综合征或伴镜下血尿,部分患者也可表现为孤立性血尿,常可伴有高血压,并可进展至肾衰竭。如患者突然发生肾衰竭,则可能因并发溶血尿毒综合征所致。据文献报道,对患者进行血清前胶原Ⅲ多肽的检测,有利于对其作出诊断。肾小球病变主要表现为系膜基质增多和毛细血管壁增厚(图6-10),可使肾小球毛细血管襻体积增大,PAS染色可显示其呈弱阳性的内皮下物,伴系膜基质插入,其形态与血栓性微血管病十分相似,或当其伴有系膜细胞增生时,酷似Ⅰ型MPGN (图6-11)。免疫荧光检查常呈阴性,或仅有局灶非特异性IgM和补体的沉积。若用抗Ⅲ型胶原的抗体进行免疫荧光或免疫组化染色,可证实其在肾小球内呈弥散性分布。电镜检查可清晣地显示,Ⅲ型胶原纤维大量分布在肾小球内皮下、系膜区(图6-12~6-14),偶沉积于GBM致密层、内外疏松层。因本病在全世界范围内报道的病例数还不多,故对其预后如何,目前仍不得而知。

▲图6-10 Ⅲ型胶原肾小球病(HE ×400)
肾小球轻度系膜基质增多伴毛细血管壁增厚

▲图6-11 Ⅲ型胶原肾小球病(HE×200)
肾小球血管襻系膜细胞轻度增生伴基质增多及血管壁增厚,酷似膜性增生性肾炎

▲图6-12 Ⅲ型胶原肾小球病(EM×15 000)
与图6-11 为同一病例,肾小球系膜区电子致密物(D)伴胶原纤维(↑)沉积

▲图6-13 Ⅲ型胶原肾小球病(EM×15 000)
与图6-10 为同一病例,肾小球内皮下区明显增厚,内有大量胶原纤维沉积

▲图6-14 Ⅲ型胶原肾小球病(EM×51 000)
与图6-10为同一病例,肾小球系膜区有多量呈周期相排列的胶原纤维沉积
六、纤连蛋白肾病
纤连蛋白肾病(fibronectin nephropathy)是一种以蛋白尿,伴进行性肾功能损害为表现的遗传性疾病。1980年,首先由Burgin等以“家族性巨纤维沉积物肾小球病”(familial glomerulopathy with giant fibrillar deposits)的名称报道,以后又有作者根据其肾小球病变特征称其为“家族性分叶状肾小球病”(familial lobular glomerulopathy)和“特发性分叶状肾小球病”(idiopathic lobular glomerulopathy)。目前,根据患者常在家族的第1代亲属中发生的现象,确认其传递方式是常染色体显性遗传。本病极为少见,据国外近20余年来的文献统计,在全世界范围内被发现的病例数还不足40例,我国至今未见其报道。本病最突出的病理改变是血浆型Fn在肾小球系膜区和内皮下的大量沉积,其沉积的前身物可能由肝脏合成,并进入血液循环所致。目前对其在肾小球内发生大量沉积的机制还不明,多数作者认为,可能与机体对其清除发生障碍与其以一种不被清除的形式存在有关,但究竟是其结构异常,还是伴有某种不被清除的蛋白质系列所致,目前还不清楚。
根据已报道病例的统计,患者多为青少年或成年人,其平均年龄为29.4岁。起病时,以蛋白尿最为常见,近半数病例可表现为肾病综合征,部分病例还可伴有镜下血尿和高血压,无血清学异常。肾组织的病理改变主要由Fn在肾小球内沉积引起,表现为肾小球体积增大,系膜区范围扩大,伴轻度系膜细胞增生和周边部GBM增厚,其沉积物经PAS、Masson染色呈强阳性,少数病例可显示血管壁呈双轨状改变和上皮下钉突形成。免疫荧光检查常呈阴性,偶有肾小球系膜区或毛细血管壁IgG、C3或伴有IgM和纤维蛋白沉积,免疫组化技术证实,肾小球系膜区呈强阳性的Fn表达。电镜检查显示,以肾小球系膜区为主的大量电子致密物沉积,也可分布于内皮下区。GBM多为正常,偶有在同一家族中发现有上皮下区和膜内沉积。沉积的电子致密物呈无定形颗粒状,可有电子透明区及散在分布、直径为10~14 nm的细丝状物,其形态介于淀粉样纤维及轻链性肾病中所见的结构模糊丝状物之间。据文献记载,本病呈进行性进展,部分病例(5/28例)经2~10年随访后,发现其已进展到慢性肾衰竭。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










