



第三节 基因打靶
基因打靶(gene targeting)又称为定向基因转移,是一种定向改变生物活体遗传信息的实验手段。通过对生物活体遗传信息的定向修饰,包括基因灭活、点突变引入、缺失突变、外源基因定位引入和染色体组大片段删除等,并使修饰后的遗传信息在生物活体内遗传,表达突变的性状,从而可以研究基因功能等生命科学的重大问题,以及提供相关的疾病治疗和新药筛选评价模型等。
一、基因打靶研究的背景
前两节提到的基因转移技术,如显微注射、电穿孔、磷酸钙沉淀及反转录病毒载体感染等方法,已能有效地将外源基因导入靶细胞内。但这些导入的外源基因在靶细胞基因组中整合的位点一般是随机的,可能导致下面几种情况出现:①导入的外源基因整合入某一正常基因的中部,导致该正常基因表达或功能的缺失;②导入的外源基因整合入细胞内正常基因的侧翼序列,影响了其周围正常基因的活性;③外源基因的导入有可能激活细胞内的原癌基因;④导入的外源基因由于其整合位点的不适,而在细胞内不表达或表达难以控制。为避免上述情况的出现,最好的途径就是将外源基因导入预先确定的位点。即对细胞基因组内靶位点进行定点修饰,也就是基因打靶,来研究该基因的功能或在生物发育中的作用。随着人类及40多种微生物基因组测序的完成,发现了大量未知功能的基因,应用基因打靶技术对这些新基因进行靶向修饰,是研究其功能最直接、最有效的手段。因此,基因打靶就日益成为继基因转移技术后亟待发展的新技术。
二、基因打靶原理
基因打靶技术是建立在ES细胞与同源重组技术基础之上的。生物界同源重组现象的发现,为基因打靶奠定了坚实的理论基础,而ES细胞技术的发展,促进了基因打靶的广泛应用。同源重组(homologous recombination)又称一般性重组或非特异性重组(general recombination),是指相似的DNA片段交换遗传信息的过程,即外源DNA片段可与宿主基因组的相应片段发生交换(即重组)。基因打靶就是用含已知序列的DNA片段与受体细胞基因组中序列相同或相近的基因发生同源重组,整合至受体细胞基因组中并得以表达或将特定基因去除来实现基因敲入或基因敲除。
ES细胞是一类具有在体外培养条件下保持未分化状态的、增殖能力强,并具有分化为多种细胞类型潜能的细胞。ES细胞注入体内与完整胚胎形成嵌合体后,可以发育形成包括生殖细胞在内的一系列成体组织。正是由于ES细胞的特殊功能,ES细胞基因打靶技术已被广泛地应用于建立转基因动物之中。
三、基因打靶的基本程序
基因打靶构建转基因动物包括3个步骤:①构建打靶载体,通过各种真核细胞转染方法将其转入ES细胞,采用正负选择法筛选已击中的细胞,或通过PCR和杂交进一步鉴定;②将中靶细胞注射入胚泡并植入假孕小鼠的子宫中;③进行表型研究和嵌合体的杂交育种(图13-5)。
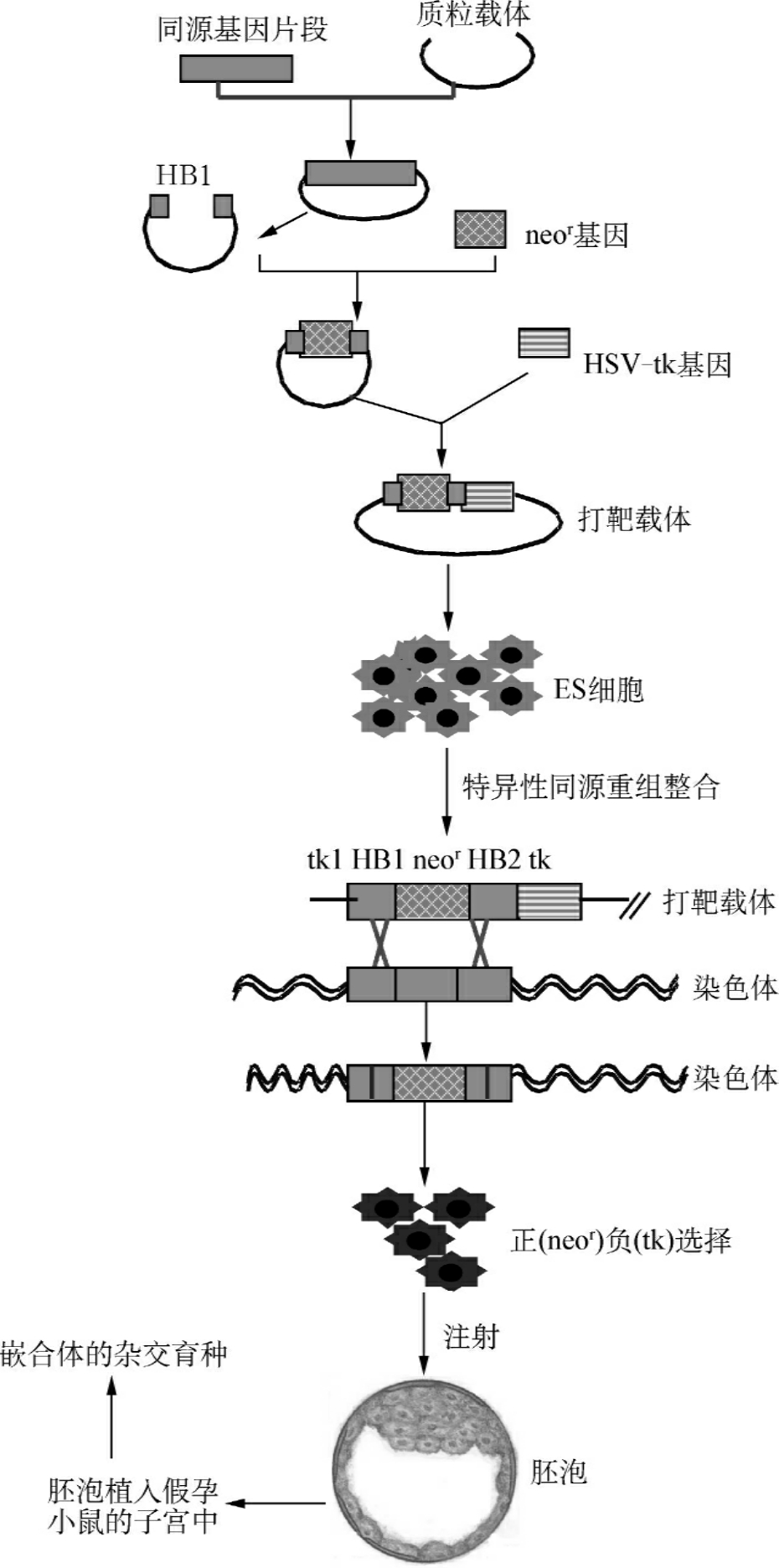
图13-5 基因打靶过程
1.基因载体的构建 基因打靶的关键是打靶载体的构建。基因打靶载体包括载体骨架、靶基因同源序列和目的序列及选择性标记基因(如neo基因,tk基因等)等非同源序列,其中同源序列是同源重组效率的关键因素。基因打靶载体有基因插入型载体(gene-insertion vector)和基因置换型载体(gene-replacement vector)。插入型载体中与靶基因同源的区段中含有特异的酶切位点,线性化后,同源重组导致基因组序列的重复,从而干扰了目标基因的功能。置换型载体进行线性化的酶切位点在引导序列和筛选基因外侧,线性化后,同源重组使染色体DNA序列为打靶载体序列替换。大多数基因敲除突变都采用置换型载体进行基因打靶。
2.ES细胞的获得 基因敲除一般采用ES细胞,最常用的是鼠,而兔、猪、鸡等的ES细胞也有使用。常用的鼠的种系是129及其杂合体,因为这类小鼠具有自发突变形成畸胎瘤和畸胎肉瘤的倾向,是基因敲除的理想实验动物。而其他遗传背景的ES细胞系也逐渐被发展应用。
3.外源DNA导入和同源重组 将重组载体通过一定的方式(如电穿孔法等)导入同源的ES细胞中,使外源DNA与ES细胞基因组中相应部分发生同源重组,将重组载体中的DNA序列整合到内源基因组中,从而得以表达。外源DNA导入的方式主要有显微注射法、电穿孔法、精子载体法、磷酸钙-DNA共沉淀法、脂质体介导法和反转录病毒法等。目前,应用最广的是显微注射法和电穿孔法。Capecchi报道,用显微注射法导入可得到很高的转染效率,占接受外源DNA细胞的10%~20%,但显微注射每次只能注射一个细胞,而电穿孔法可同时使许多细胞得到转染。Mansour等研究发现电穿孔可使1%的ES细胞稳定转染。
4.筛选已击中的细胞 由于基因转移的同源重组自然发生率极低,动物的重组概率为10-2~10-5,植物的概率为10-4~10-5。因此,如何从众多细胞中筛出真正发生了同源重组的ES细胞非常重要。目前,常用的方法有正负筛选法(positive negative selection,PNS法)、标记基因的特异位点表达法以及PCR法。其中应用最多的是PNS法,其基本原理是:同源重组时,只有载体的同源区以内部分发生重组,同源区以外部分将被切除。随机整合时,是在载体的两端将整个载体连入染色体内。置换型载体含有正负选择基因各一,正选择基因多为新霉素磷酸转移酶(neo基因),位于同源区内,其在随机整合和同源重组中均可正常表达。负选择基因在靶基因同源区之外,位于载体的3′端,常用单纯疱疹病毒胸腺嘧啶激酶(HSV-tk基因),在同源重组时tk基因将被切除而丢失,相反在随机整合时,所有的序列均保留(包括胸苷激酶,tk)。tk可使无毒的丙氧鸟苷(GANC)转变为毒性核苷酸,而杀死细胞,因而可用丙氧鸟苷筛选排除随机整合的细胞株。故同源重组时,细胞对G418和GANC都有抗性,随机整合时对G418有抗性,但对GANC敏感,细胞将被杀死,无整合的将被G418杀死。用G418作为正筛选,选出含有neo基因的细胞株,再用GANC作为负筛选淘汰含有tk基因的细胞株,保留未含有tk基因的同源重组细胞株,此方法是目前应用较广泛的一种策略。正向筛选标记基因neo具有双重作用,一方面导致靶基因的插入突变;同时作为重组细胞的正向筛选标志,该基因产物可使细胞在含有新霉素的培养基中生长。
5.表型研究和育种 通过观察嵌合体小鼠的生物学性状,进而了解目的基因变化前后小鼠性状的改变,达到研究目的基因的目的。由于同源重组常发生在一对染色体中的一条染色体上,所以如果要得到稳定遗传的纯合体基因敲除模型,需要进行至少两代遗传。
四、基因打靶的策略
1.完全基因剔除(complete knock out) 在ES细胞中进行基因打靶最常用的策略依然是使用正负选择载体。将阳性选择标记基因插入到靶基因功能最关键的外显子中,或通过同源重组删除靶基因最重要的功能域,实行靶基因的完全剔除。阳性选择标记基因常采用基因neo或加上报告基因β-半乳糖苷酶基因(lacZ),将它们以正确的读框插入到靶基因的外显子中,除了剔除靶基因外,还可通过分析β-半乳糖苷酶的活性,研究靶基因的时空表达情况。
此外,由于阴性选择基因(多为tk基因)可能在转染及整合过程中失活,导致打靶载体随机整合的ES细胞克隆得以生长,因而PNS载体依然得到较多随机整合的ES细胞克隆。为了提高中靶细胞筛选的效率,常采用启动子缺失的打靶载体。载体中阳性选择标记基因neo是不带启动子的,只有发生正确的同源重组,neo基因被精确地插入靶基因启动子后,抗性基因才会表达,并使ES细胞克隆在选择性药物培养基中存活下来。若在neo基因和lacZ基因间连接上内源核糖体插入位点(internal ribosomal entry sites,IREs),则可让neo基因和lacZ基因在一次转录中同时被翻译。
2.随机基因剔除—基因捕获 1989年,Gossler等首先应用一种极有创意的方法,在ES细胞中使用随机插入突变技术进行基因诱变,并形象地命名为“基因诱捕”。它的基本原理是通过物理、化学和生物学等方法将一个带有外源基因,如抗药基因或报告基因的DNA载体导入到ES细胞中,使其随机整合到基因组中。导入的抗药基因,通常是neo基因,一般无启动子,neo基因插入到ES细胞染色体组后利用捕获基因的转录调控元件实现表达,使ES细胞克隆可以在含G418的选择培养基中筛选出来。
用基因捕获法在单次实验中可以获得数以百计带有单基因剔除的ES细胞克隆。但此方法的缺点是只能剔除在ES细胞中表达的基因。随着应用哺乳动物体细胞克隆新个体的成功实现。在体细胞中应用基因捕获剔除更多在发育晚期才表达的基因,其后通过核移植技术获得带有突变基因的动物个体、这不失为一种可以弥补以上缺陷的办法。此外,用基因捕获法进行基因剔除的另一个缺点是无法对基因进行精细的遗传修饰。
3.精细突变的引入 人类疾病中许多是由于基因功能丧失引起的,也有许多是由于基因过度表达或功能获得引起的。对后者就无法用基因剔除的方法获得相应的疾病模型。为此,研究者发明了各种可以将诸如插入终止密码子或替换某个氨基酸之类的精细突变引入小鼠基因组中的方法。早期的研究者采用的方法主要有两种:①将不含选择标记基因的打靶载体直接经显微注射法引入ES细胞,然后用PCR分析鉴定同源重组克隆;②是用带有精细突变但无选择标记基因的打靶载体与仅含标记基因的载体经电穿孔共转染ES细胞。这两种方法在技术上有很大局限性,现在基本上已不再使用。目前,较为常用的方法主要有“Hit and Run”、“Tag and Exchange”以及基于重组酶系统的方法。
(1)打了就走策略(Hit and Run法):也称进退策略,这一方法包括两次同源重组。第一次(Hit):用线性化插入型载体进行同源重组,这样会在靶位点插入带有突变的基因组序列以及载体骨架,载体骨架上带有两个筛选标记(如正筛选标记基因neo和负筛选标记基因tk),进行正选择。第二次(Run):再通过染色体内重组,去除了含有负筛选标记基因的载体序列,由于负筛选标记基因的消失,细胞就恢复了对负筛选药物的抗性,利用tk基因抗性筛选,富集为数不多的发生了染色体内重组的克隆。这样就在基因组中引入突变基因而不引入筛选标记基因。
(2)双置换法(Double Replacement):最早提出这一设想的研究者称其为“In-Out”法。1995年,Moore等进行了改进,称其为“双置换法”。第一步:用含有次黄嘌呤磷酸核糖转移酶(Hprt)基因的打靶载体转染Hprt-ES细胞,Hprt基因双侧是靶基因的同源序列以及tk基因。通过在次黄嘌呤(HAT)和GANC培养基中筛选发生同源重组、Hprt基因整合到基因组中的阳性克隆。第二步:用只携带突变同源序列的打靶载体转染第一步获得的Hprt+ES细胞。同源重组发生后,突变序列整合入基因组,Hprt被置换出来。Hprt-ES细胞可在6-GT培养基上筛选并用PCR进行基因组分析。
(3)“标记和置换”法(Tag and Exchange):策略与双置换法有许多共同之处。它是用两个不同的置换型载体进行两次连续的基因打靶完成的。通过第一步的同源重组插入正负筛选标记基因(如neo和tk)对靶基因进行“标记”。第二步的打靶载体的同源序列中间带有精细突变的序列,该载体转染第一步得到的ES细胞后,带有精细突变的同源序列将置换下被“标记”的靶基因,从而将neo和tk基因置换下来,用GANC筛选带有靶基因精细突变的细胞。
(4)利用Cre-LoxP系统引入点突变:近年来,导入精细突变最常用的方法应首推以Cre-loxP为基础的重组系统。Cre(引致重组)基因表达产物是位于特异性重组发生所需的特异性重组酶,其识别位点为loxP(交换基因座,locus of crossingover),为一段34bp的DNA序列,由2个13bp的反向重复序列和1个8bp的不对称间隔序列组成。应用Cre-loxP系统将精细突变导入基因组的策略为:在置换型的打靶载体中,正负筛选标记基因两侧各放置一个loxP序列,并被置于靶基因的内含子中。携带精细突变的外显子位于载体一侧的同源臂上。经过同源重组筛选和突变的鉴定(如利用新产生的酶切位点来鉴定)后,将Cre重组酶表达质粒导入中靶ES细胞。Cre介导的位点特异性重组再将两个loxP之间的正负筛选标记基因切除,最后通过GANC培养基来富集带精细突变目的基因的中靶细胞。
4.染色体组大片段的删除和重排
(1)Cre介导的非同源染色体间的删除:基于Cre-loxP系统的位点特异性重组可以介导长距离的重组来实现大片段的删除。具体的实施过程有两种策略:①通过2次打靶分别将2个loxP位点引入靶位点,中靶的克隆转染表达Cre酶的质粒,去除2个loxP位点之间的序列;②第1次中靶的克隆直接转染第2次的打靶载体和Cre酶表达质粒,直接筛选最终中靶的克隆。
(2)Cre-loxP介导的姐妹染色体间的重排:哺乳动物细胞中有相当数量的基因是多拷贝的,这些多拷贝的基因在染色体上排列在一起形成基因簇。基因簇中的基因具有相同或相似的功能。用基因打靶的方法研究这些基因的功能必须使这些基因同时产生突变。如果基因簇内的各基因的遗传距离较大,可以分别在每一个拷贝上引入突变,通过子代的杂交获得每个拷贝同时带有突变的个体。但当基因簇内的各基因间的距离很小时,染色体交换的频率非常低,无法通过子代杂交获得各拷贝同时带有突变的个体。用Cre-loxP系统可以实现这一目的。具体的实施策略是将2个loxP序列通过同源重组分别引入到2个基因中去,得到2个分别带有loxP序列的小鼠品系。2个品系杂交,就会得到在2条姐妹染色体上同时带有loxP序列的小鼠,再和表达Cre酶的第3个小鼠品系杂交便可获得2个拷贝同时删除的子代。
(3)Cre介导的非同源染色体间的重排:人类的许多遗传疾病是由于染色体的易位引起的,应用Cre-loxP系统成功地建立了相应的动物模型。其原理是:将loxP序列分别引入到非同源染色体上,在Cre酶的存在下诱导非同源染色体之间的重组。
5.条件性基因打靶 基因打靶技术尽管克服了经受精卵原核显微注射技术引起的,诸如外源基因随机整合,其拷贝数不能控制的局限性,但由于基因敲除小鼠的所有细胞基因组上都存在基因的缺失/突变,往往引起严重的发育缺陷或胎儿死亡,不利于在发育后期阶段基因功能的分析。即使发育完整的突变体小鼠,对于基因敲除表型的解释也常会遇到2个困难问题:①所有体细胞基因的剔除,很难将异常的表型归于哪一类细胞或组织;②很难排除在成熟动物上由于发育缺陷所引起的异常表型。另外,由于广泛应用neo和tk作为正负选择系统,发生同源重组的细胞基因组中总留有外源的选择标记(neo)基因,该基因可能影响相邻基因的表达,不利于对突变表型的精确分析。以Cre/loxP系统与基因打靶技术相结合的条件性基因打靶将可克服上述的局限性。该策略主要是运用打靶基因置于同向loxP之内的打靶载体,经在ES细胞上同源重组及筛选,将携带该载体的小鼠与受控于组织特异性启动子或诱导性启动子的Cre基因的TgM交配,经Cre介导重组,即可获得靶基因在某一组织器官或发育时期上缺失的基因敲除小鼠。条件基因打靶有以下特点:①通过条件性基因敲除,研究完全敲除具有致死效应的基因的功能以及基因在特定的组织细胞或个体发育特定阶段的功能;②通过条件性基因激活,实现转基因的可控制性表达;③通过Cre切除条件性基因修复(Cre excision-conditional gene repair),进行基因的可修复性敲除,以研究一个基因的多种功能。
应用Cre-loxP系统,研究者可以如愿以偿地在不同的时相、不同的空间按预期的设计进行基因剔除。然而,现阶段可利用的组织特异性表达Cre重组酶的转基因小鼠还十分有限。对Cre转基因表达的精确控制还依赖于更多组织特异性标志基因的发现以及人工调控基因表达系统的进一步研究。
五、RNAi引起的基因敲除
1995年,康奈尔大学的Su Guo博士用反义RNA阻断线虫基因表达的实验中发现,反义和正义RNA都阻断了基因的表达,他们对这个结果百思不得其解。直到1998年,Andrew Fire的研究证明,在正义RNA也阻断了基因表达的实验中,真正起作用的是ds RNA。这些ds RNA是体外转录正义RNA时生成的,称为siRNA。这种ds RNA对基因表达的阻断作用被称为RNAi。随后的研究中发现,RNAi现象广泛存在于线虫、果蝇、斑马鱼、真菌以及植物等生物体内,这些生物体利用RNAi来抵御病毒的感染,阻断转座子的作用。RNAi能高效特异的阻断基因的表达,在线虫和果蝇体内,RNAi能达到基因敲除的效果。在小鼠和人的体外培养细胞中利用RNAi技术也成功阻断了基因的表达,实现了细胞水平的基因敲除。近年来,越来越多的基因敲除采用了RNAi这种更为简单方便的方法。
1.RNAi阻断基因表达的机制 ds RNA进入细胞后,能够在Dicer酶的作用下被裂解成siRNA,而另一方面ds RNA还能在RdRP(以RNA为模板指导RNA合成的聚合酶:RNA-directed RNA polymerase)的作用下自身扩增后,再被Dicer酶裂解成siRNA。siRNA的双链解开变成单链,并和某些蛋白形成复合物,Argonaute2是目前已知的参与复合物形成的蛋白。此复合物同与siRNA互补的mRNA结合,一方面使mRNA被RNA酶裂解;另一方面以siRNA作为引物,以mRNA为模板,在RdRP作用下合成出mRNA的互补链。结果mRNA也变成了ds RNA,它在Dicer酶的作用下也被裂解成siRNA。这些新生成的siRNA也具有诱发RNAi的作用,通过PCR反应,细胞内的siRNA大大增加,显著增加了对基因表达的抑制。从21~23个核苷酸的siRNA到几百个核苷酸的双链RNA都能诱发RNAi,但长的ds RNA阻断基因表达的效果明显强于短的ds RNA。
RNAi机制被发现以来,在低等模式生物中得到了广泛的应用。然而在哺乳动物中,导入长度>30bp的ds RNA就会启动哺乳动物体内的抗病毒保护机制,并且没有特异的靶向性。Elba Shir等发现将人工合成的21~23bp大小的ds RNA导入哺乳动物细胞后,可以引起特异的基因沉默,并不会引发细胞的抵御反应。不足之处就是这种特异的基因沉默效果不能长期维持。2002年,多家实验室几乎同时将利用RNA聚合酶Ⅲ的启动子构建的RNAi载体应用于转染哺乳动物的细胞系,从而达到稳定的对靶基因产生转录后沉默的效果。同时,Ma saru Okabe实验室成功建立了第1种应用RNAi的转基因小鼠。由于RNAi转基因小鼠模型能够达到类似基因敲除的效果,迄今为止,已经有几百篇关于RNAi转基因小鼠的报道。引导siRNA产生的启动子,已经由RNA聚合酶Ⅲ的H1和U6扩展到了RNA聚合酶Ⅱ。对siRNA的设计要求也由原来的21~23bp扩展到了几百个碱基。基于四环素等诱导的条件型RNAi转基因策略的出现,使得这一新的研究基因功能的强有力的工具受到越来越多的关注。
2.ds RNA的构建 ds RNA可先在体外构建好,然后转染细胞。在体外构建ds RNA时,分别在体外转录出正义和反义RNA,再将两者退火,形成ds RNA。体外合成的ds RNA可以用脂质体转入细胞中。但有些细胞脂质体转移效果差,转移到细胞内的ds RNA半衰期短。而先在体外构建能表达ds RNA的载体,再将载体转移到细胞内,在细胞内合成出ds RNA,不但能增加有效转染细胞的种类,而且在长期稳定表达载体的细胞株中,ds RNA能够长期发挥阻断基因的作用。
在构建ds RNA的表达载体时,使用RNA聚合酶Ⅲ来指导RNA的合成。U6启动子能被RNA聚合酶Ⅲ识别,合成出RNA。RNA聚合酶Ⅲ还能识别H1-RNA启动子。在H1-RNA启动子后面接上能形成发卡样结构的反向互补序列,将此载体转入细胞后也能在细胞内合成dsRNA。T7也可作为启动子合成dsRNA。将PCR产物用NotⅠ酶切后,回收正向片段和反向片段连接成的具有反转重复序列的片段,接到pGEMTeasy载体上,就构建成了可以表达dsRNA的载体。
腺病毒是体内转基因的常用载体。用腺病毒做载体在体内和体外表达dsRNA已取得成功,并成功地阻断了基因的表达,从而实现了成体动物的基因敲除。
3.RNAi基因敲除的优点 ①RNAi基因敲除比用同源重组法更加简便,周期大大缩短;②对于哺乳动物,如对于一些敲除后小鼠在胚胎时就会死亡的基因,可以在体外培养的细胞中利用RNAi技术研究它的功能;③可以在一个转基因动物体内同时沉默两种或更多基因,加快了获得突变的转基因动物的进程;④通过制备针对靶基因mRNA不同区段的siRNA转基因动物,有可能获得对靶基因不同程度的沉默效果,从而可以模拟数量遗传性状。
六、基因打靶的影响因素
基因打靶的效率是指可检测到的细胞中发生同源重组的细胞数与转染的细胞总数之比,又称同源重组效率。基因打靶效率在不同的研究报道中变动范围较大,其波动性来自较多的影响因素。
1.打靶载体的影响 打靶载体中同源序列是决定同源重组效率的关键因素,Thomas和Capecchi的研究表明,当载体同源序列长度从4kb增至9kb时,基因打靶效率增加10倍,与此同时,非同源重组效率增加40倍。但同源序列达一定长度后,继续增加长度对同源重组效率不产生显著影响。另外,载体上的非同源序列对打靶效率可能产生影响,片段插入的数目和位点可能与同源重组效率有关,但不是主要的影响因素。
2.外源DNA导入方式的影响 目前,外源DNA导入的方式主要有显微注射法、电穿孔法、精子载体法和反转录病毒感染法。不同的导入方法对同源重组效率有明显影响。目前,应用最广的是显微注射法,用显微注射法导入可得到很高的转染率,占接受外源DNA细胞的10%~20%,但显微注射每次只能注射1个细胞,而用电穿孔法可同时使许多细胞得到转染,可使1%的ES细胞稳定转染。小鼠ES细胞的建立,为基因打靶提供了受体细胞,ES细胞传代稳定,体外可操作性强,可在体外培养的水平上进行转基因整合的筛选鉴定,达到定点整合的效果,对制作转基因小鼠和研究转基因的结构和功能起到了巨大的推动作用。近年,精子载体法研究也在不断地进行着,如何使DNA与精子结合可能是获得成功的一个关键因素。而反转录病毒载体法利用某些病毒对某些组织具有特异的亲和力,在人类疾病的基因治疗方面显示了良好的发展潜力。
3.基因的影响 早期研究缺陷型标记基因时发现整合标记基因的同源重组效率与它在基因组中的插入位置有关,说明基因组中的不同区域对同源重组过程产生了影响。Thomas和Doetschman等在小鼠ES细胞Hprt基因的定点突变中,以基因顺序的不同部分作为目的突变区域,结果发现同源重组效率有明显差异,说明基因结构顺序对同源重组效率有影响。一般认为,转录活性基因有利于同源重组,靶基因拷贝数的增加并不能提高同源重组效率。
4.调控元件的影响 目前,转基因的整合具有位置效应已被公认,由于受整合位点周围染色质翼区的影响,因而在许多情况下影响到表达水平和组织特异性表达,5′端的启动子和增强子,3′端终止子都对转基因的整合产生作用,这也是某些打靶载体选择组织特异性启动子的原因。外源转基因的有效表达还取决于其是否有内含子,因为内含子中存在的转录调控元件可影响mRNA的剪接和启动子与内含子间的调节反应。另外,内含子可能含有能开放染色体功能域的一些序列,也可能通过影响核质成分、位置等来提高转基因表达。此外,目的基因插入片段大小、载体DNA是否线性化等也影响基因打靶的效率。
七、基因打靶技术的应用前景和展望
1.基因功能的研究 后基因组时代主要任务就是研究大量新基因的功能。用体细胞基因打靶技术通过定点改造基因组中的特定基因,有可能在细胞水平上研究某一基因的功能及调控机制;从定点突变的干细胞获得基因突变型个体,可在生物体整体水平上了解某些基因在体内的具体作用。另外,胚胎发育是非常复杂的生命现象,在这一过程中包含着许多生理、生化的复杂变化,尤其是要考察某一基因对某一组织器官发育的影响,用传统的研究方法很难进行观察研究。基因打靶为这一领域的研究提供了理想的方法。
2.建立人类疾病的动物模型 人类疾病动物模型对病理研究及临床治疗非常重要。自发或诱变病理模型需漫长的时间;应用转基因技术,外源基因在基因组中的随机整合可能带来不确定的表型。基因打靶技术在很大程度上克服了上述不足。通过对ES细胞打靶可获得含特定突变基因的小鼠模型。
3.用于疾病的基因治疗 通过基因敲入技术将正常基因引入到病变细胞中,取代原来异常的基因或对缺陷基因进行精确改正,使修复后的细胞表达正常蛋白,不再表达错误产物,是一种理想的基因治疗策略。另外,可通过基因敲除技术敲除多余的、过量表达的及影响正常生理功能的基因以达到治疗目的。
4.用于改造生物和培育新的生物品种 基因打靶技术的出现使转基因动物和生物反应器的制备更为精确,外源基因将被准确地插入受体的基因组中,定点改造原有的基因功能,使生物获得优良的性状,并避免由于外源基因在基因组中随机整合可能带来的不利影响。对动物生殖细胞,或早期ES细胞的基因进行修饰改造可以产生一些人类需要的新品种。
5.基因打靶技术的缺陷 随着基因打靶技术的发展,早期技术中的许多不足和缺陷都已经解决,但基因打靶技术始终存在着一个难以克服的缺点,即敲掉一个基因并不一定就能获知该基因的功能,其原因包括:①许多基因在功能上是冗余的,敲掉一个在功能上冗余的基因,并不能造成容易识别的表型,因为基因家族的其他成员可以提供同样的功能;②对于某些必需基因,敲除后会造成细胞的致死,也就无法对这些必需基因进行相应的研究了。
(潘銮凤)
1.陈永福主编.转基因动物.北京:科学出版社,2002
2.杨晓,黄培堂,黄翠芬主编.基因打靶技术.北京:科学出版社,2003
3.周兴,张居农.精子载体转基因技术研究进展.中国奶牛,2007,8:27~31
4.郭蓉,吴登俊.精子载体法在转基因动物中的应用与前景.当代畜牧,2006,4:43~46
5.乔永等.精子介导的转基因技术研究进.畜牧与兽医,2006,38(7):57~50
6.信吉阁等.RNAi及其在哺乳动物中的应用.云南农业大学学报,2006,21(2):210~216
7.尹秀山,张令强,贺福初.RNAi技术在转基因动物中的应用.遗传,2006,28(3):351~356
8.张定校等.RNA干涉(RNAi)技术应用于哺乳动物细胞的研究策略.遗传,2005,27(5):839~844
9.李若楠,张彦才.转基因动物技术及其应用.华北农学报,2007,22(增刊):181~184
10.王锋,蒋晓明.小鼠体细胞核移植程序的研究.四川动物,2006,25(3):459~562
11.滕艳,杨晓.基因打靶技术:开启遗传学新纪元.遗传,2007,29(11):1291~1298
12.万海英,汤华.基因敲除技术现状及应用.医学分子生物学杂志,2007,4(1):86~90
13.杜娟,罗桂芬.胚胎干细胞途径的基因打靶技术.陕西农业科学,2006,5:70~73
14.张传生,杜立新.基于Cre重组酶体系的鸡卵清蛋白基因打靶载体的构建.动物学报,2005,51(4):685~690
15.李涛,卢圣栋.时空特异性基因打靶的应用及有关进展.生物工程进展,2002,22(2):56~60
16.Lavitrano M,et al.Sperm cells of vectors for introducing forgeign DNA into eggs:genetic transformation of mice.Cell,1989,57:717~723
17.Gordon,Gordon JW,et al.Genetic transformation of mouse embryo by microinjection of purified DNA.Proc Natl Acad Sci USA,1980,77:7380~7384
18.Brinster RL,et al.Regulation of metallothionein-thymidine kinase fusion plasmids injected into mouse eggs.Nature,1982,296:39~42
19.Thomson JA,et al.Embryonic stem cell lines derived from human blastocysts.Science,1998,282(5391):1145~1147
20.Gossler A,et al.Mouse embryonic stem cell and receptor constructs to detedct developmentally regulated gene.Science,1989,244(4903):463~465
21.Moore RC,et al.Double replacement gene targeting for the production of a series of mouse strains with different prion protein gene alterations.Biotechnology,1995,13(9):999~1004
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。











