



第二节 基因诊断的方法选择
人类疾病的发生主要是由于内源基因的改变和外源基因的入侵。与疾病相关的内源遗传背景改变主要有两类:①遗传物质,即DNA或RNA的水平的变化。如病毒感染量、病毒基因及其转录产物在人体内的从无到有,某些肿瘤中癌基因表达水平的从低到高。②遗传物质的结构变化,即基因突变。如点突变引起的基因失活及染色体转位引起的基因激活或灭活等。所以,针对这些病因可采取相应的基因诊断方法,主要包括以下几个方面。
一、基因结构改变的诊断
基因结构的改变包括点突变、核苷酸片段插入或缺失,基因重排或易位,基因扩增,前病毒插入等。根据不同的基因改变情况来选择不同的方法。
(一)点突变和少数核苷酸插入或缺失的诊断
1.已知突变 对于突变基因及突变位点已经清楚的点突变或少数核苷酸插入或缺失,可采用PCR结合ASO探针杂交分析进行检测(allele specific oligonucleotide,ASO)。ASO探针:等位基因特异性寡核苷酸探针,指针对各种正常和突变靶序列设计的特异性寡核苷酸探针。寡核苷酸中的碱基错配会大大影响杂交分子的稳定性,因此可用人工合成的针对正常和突变等位基因的特异性寡核苷酸探针进行杂交,检测点突变。
首先在突变位点两端设计一对引物进行PCR扩增,然后可采用两种方法进行杂交:①将PCR产物变性后斑点印迹至膜上,用一系列ASO探针杂交,严格控制杂交和洗膜条件、确保正常ASO探针只与正常靶序列杂交,而突变ASO只与含相应突变碱基的靶序列杂交;②将ASO探针固定在膜上,然后与生物素或地高辛标记的PCR产物杂交,即反向斑点杂交(reverse dot-blot),这样一次杂交可完成多个探针检测。
另外还可采用等位基因特异性扩增(alleles specific amplification,ASA),又称扩增阻碍突变系统(amplification refractory mutation system,ARMS)来检测已知点突变。也就是利用PCR引物的3′端末位碱基必须与其模板DNA互补才能有效扩增的原理,设计等位基因特异性的PCR扩增引物,在严格的条件下,只有在引物3′碱基与模板配对时才能出现PCR扩增带,从而检测出突变,该法省去了探针杂交操作。
当待测DNA序列中发生突变时会导致某些限制性内切酶位点的改变,其特异的限制性酶切片段的状态在电泳迁移率上也会随之改变,借此可作出分析诊断。其方法有两种:①根据已知的变异选用特定的限制性内切酶酶切DNA,然后用特异探针进行Southern杂交,根据杂交条带的变化作出诊断;②用PCR法扩增基因片段,再用特定限制性内切酶酶切扩增产物,根据电泳条带的大小判断是否有异常。
2.未知突变 寡聚核苷酸探针分析法一般应用于已知序列的点突变疾病基因的检测。对于那些是否发生变异的基因或突变位点或性质未知的突变则可采用PCR/SSCP/测序的方法。通过PCR扩增待测基因和野生对照基因的DNA片段,将扩增产物变性成单链,进行SSCP分析,若两者的迁移率不同,即可能发生突变,然后通过测序得知确切的突变。
DNA芯片技术可用于大规模未知突变的筛查,比如可针对一段4kb的序列内每一个核苷酸设计4个探针,共16000个寡核苷酸探针,通过一次杂交即可确定所有突变的性质和位点。对于每一个位点的核苷酸设计的寡核苷酸探针分别为A、T、C或G。如果正常基因该位点为C,即与G点杂交;若与A点杂交,即为C→T的突变,依此类推,可发现C→G和C→A的突变。其他位点也照样检测,结果即可测出所有突变。
(二)大片段核苷酸插入或缺失的诊断
对于少数核苷酸的插入或缺失可采用检测点突变的PCR/杂交方法。对于插入或缺失的片段在0.5~1.5kb左右的可设计两对引物来检测。引物1和2在片段的邻近外侧两端,通过其扩增产物来确定有无缺失或缺失片段的相对大小。引物3和4设计在缺失片段的内部,来确定缺失部位(图14-2)。

图14-2 0.5~1.5kb左右片段插入或缺失的PCR检测
但PCR扩增2kb以上的片段比较困难,因此需采用多重PCR(multiplex PCR),即在同一个PCR体系中加入多对引物,扩增同一模板的几个区域(图14-3)。多重PCR引物设计的注意点:①产物大小易于分离;②为了具备较高特异性,引物长23~28nt;③所有引物的G+C含量相近,55%左右;④优化PCR条件,以适应多对引物扩增的要求。
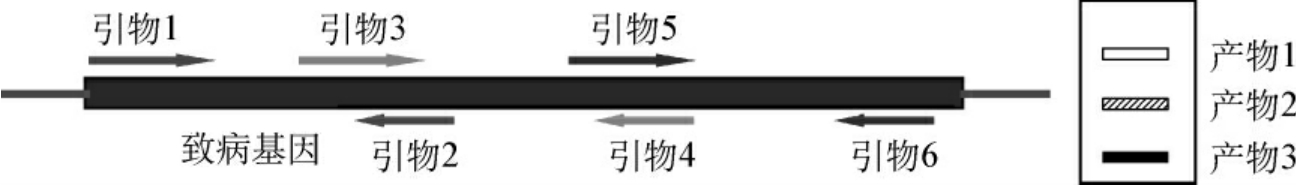
图14-3 多重PCR检测
采用多重PCR进行检测的一个典型例子为杜氏肌营养不良症(DMD)。DMD是一种性连锁隐性遗传病,是有抗肌萎缩蛋白基因决定的。该基因全长2000~2500kb,包含79个外显子,是人体中最为庞大的基因之一。该基因突变所致的抗肌萎缩蛋白的缺失或结构异常是导致发病的根本原因。抗肌萎缩蛋白基因具有较高突变率,主要表现为:约60%缺失突变,5%重复突变,35%小片段缺失或点突变。突变的热点区在基因的中央部第45~55外显子和5′区。应用多重PCR,设计针对外显子4、8、12、17、19、44、45、48、51位点的9对引物,可检出90%具有基因缺失的病例。
(三)基因重排或染色体易位的诊断
对于已知基因的重排或染色体易位可采用染色体荧光原位杂交(FISH)的方法。也可设计3条PCR引物进行2个扩增来检测,引物1与未重排或易位的正常位点的序列互补,引物2与重排或易位后的邻近位点序列互补,引物3与移动的基因的内部序列互补。引物2和3的PCR产物的有无可判断基因重排或易位与否。引物1和3的PCR产物的存在为未重排或易位的正常情况,可作为阴性对照(图14-4)。例如慢性粒细胞性白血病的易位{t(9;22)}融合了bcr和abl基因,导致了发病。

图14-4 基因重排或染色体易位的诊断
(四)基因数目改变的诊断
在一些低等真核生物的生长发育过程中,可以通过丢失某些基因而达到使这些基因永久性关闭的目的。另一方面,机体为了生长发育的需要或为了适应环境的变化,使某些基因的拷贝数大量增加,称为基因扩增。基因扩增也发现于某些类型的肿瘤中,可以从几十至上千倍不等。以待检基因DNA或cDNA做探针,采用适当的限制性内切酶把基因组DNA酶切后电泳,通过Southern印迹杂交,可以对基因扩增进行检测。如果基因组中该基因有扩增发生,杂交后显示的条带位置会发生改变,杂交条带的信号强度也会增强,对条带进行光密度扫描,采用单拷贝基因进行对照,可进行基因扩增的定量分析。
二、多态性分析
多态性(polymorphism)是指处于随机婚配的群体中,同一基因位点可存在两种以上的基因型。在人群中,个体间基因的核苷酸序列存在着差异性称为基因(或DNA)的多态性(gene polymorphism)。这种多态性可以分为两类,即DNA位点多态性(site polymorphism)和长度多态性(length polymorphism)。
1.位点多态性 是由于等位基因之间在特定的位点上DNA序列存在差异,也就是基因组中散在的碱基的不同,包括点突变(转换和颠换)、单个碱基的置换、缺失和插入。突变是基因多态性的一种特殊形式,单个碱基的置换又称为单核苷酸多态性(single nucleotide polymorphism,SNP),SNP通常是一种二等位基因(biallelic)或二态的变异。据估计,单碱基变异的频率在1/1000~2/1000。SNP在基因组中数量巨大,分布频密,在整个人类基因组中的分布可以达到300万个,在人群中的分布数目更高,检测易于自动化和批量化,被认为是新一代的遗传标记。
2.长度多态性 共有两类:一类为可变数目串联重复序列(variable number of tandem repeats,VNTR),它是由于相同的重复顺序重复次数不同所致,其典型代表有小卫星DNA(minisatellite)和微卫星DNA(microsatellite)。小卫星是由15~65bp的基本单位串联而成,重复次数在人群中是高度变异的。微卫星DNA为更短的重复序列,基本序列只有1~8bp,如(TA)n及(CGG)n等,重复次数在不同基因型间差异很大,从而形成其座位的多态性。这些短串联重复序列(short tandem repeat,STR)长度多态性是按照孟德尔方式遗传的,它们在基因定位、DNA指纹分析、遗传病的分析和诊断中广泛地应用。另一类长度多态性是由于基因的某一片段的缺失或插入所致。
3.基因多态性的医学意义 人类基因多态性在阐明人体对疾病、毒物的易感性与耐受性,疾病临床表现的多样性,以及对药物治疗的反应性上都起着重要的作用。临床上早期有关基因多态性的研究是从人类白细胞抗原(HLA)基因开始的,分析基因型在疾病发生易感性方面的作用,如HLA-B27等位基因与强直性脊椎炎发生率的密切关联,可作为诊断的依据。通过基因多态性的研究,可从基因水平揭示人类不同个体间生物活性物质的功能及效应存在着差异的本质。通过对基因多态性与疾病的易感性的联系研究,如p53抑癌基因多态性与肿瘤发生及转移的关系研究,可阐明人体对疾病、毒物和应激的易感性,不仅为临床医学也为预防医学的发展带来新的领域。
基因多态性的研究对于遗传病具有双重意义,一方面基因的有害突变,不论是经典的点突变,还是动态突变,其本身就可能是遗传病的病因。另一方面,众多的多态性位点又是很好的遗传标记,可以在遗传病的研究和临床诊断中发挥重要的作用。①多态性作为遗传病的病因,点突变引起的疾病:从镰刀状细胞贫血开始,突变引起各种遗传病的例子愈来愈多,遗传性肿瘤也逐渐被认识;②多态性作为遗传标记的应用:绝大多数DNA多态性并不引起遗传病,但可作为遗传标记来使用。例如:上述提到的各种多态性标记,包括限制性片段长度多态性(restriction fragment length polymorphism,RFLP)位点,微卫星和小卫星DNA标记都已广泛用于遗传病的连锁诊断。利用各条染色体上位置已知的众多的多态性标记,通过患病家系的连锁分析,可以找到多基因病的易感基因或相关基因的位置,并为他们的分离克隆提供依据。
4.基因多态性的主要检测方法
(1)RFLP:由DNA的多态性,致使DNA分子的限制酶切位点及数目发生改变,用限制酶切割基因组时,所产生的片段数目和每个片段的长度就不同,即所谓的RFLP,导致限制片段长度发生改变的酶切位点,又称为多态性位点。最早是用Southern blot/RFLP方法检测,后来采用PCR与限制酶酶切相结合的方法。现在多采用PCR-RFLP法进行基因限制性片段长度多态性的研究。
对于基因结构变异已经明确的疾病可采用直接分析法,如Lener遗传性视神经病,是由线粒体DNA(mtDNA)发生突变引起的。大多数Lener患者mtDNA的第11778位的G→A,这种转换使SfaNⅠ酶切位点丧失。在该位点两侧设计一对引物,可扩增出340bp的产物,未突变的正常个体的PCR产物经SfaNⅠ酶切后,出现190bp和150bp的两条带,而突变的患者只出现340bp一条带。若出现340bp、190bp和150bp 3条带,说明存在野生型和突变型两种类型的mtDNA,为杂合子个体。
RFLP按照孟德尔方式遗传。在某一特定的家系中,如果某一致病基因与特异的多态性片段紧密连锁,就可以用这一种“遗传标记”来判断家系成员或胎儿的基因组中是否带有致病基因。这种通过对RFLP的连锁分析对疾病基因进行间接诊断的方法称为RFLP间接分析法。
(2)扩增片段长度多态性(amplification fragment length polymorphism,AFLP):是一项新的分子标记技术,是基于PCR技术扩增基因组DNA限制性片段,基因组DNA先用限制性内切酶切割,然后将双链接头连接到DNA片段的末端,接头序列和相邻的限制性位点序列,作为引物结合位点。限制性片段用两种酶切割产生,一种是罕见切割酶;另一种是常用切割酶。它结合了RFLP和PCR技术特点,具有RFLP技术的可靠性和PCR技术的高效性。由于AFLP扩增可使某一品种出现特定的DNA谱带,而在另一品种中可能无此谱带产生。因此,这种通过引物诱导及DNA扩增后得到的DNA多态性可作为一种分子标记。AFLP可在一次单个反应中检测到大量的片段,说明AFLP技术是一种新的而且有很大功能的DNA指纹技术。
(3)变性梯度凝胶电泳法(denaturing gradient gel electrophoresis,DGGE):分析PCR产物,如果突变发生在最先解链的DNA区域,检出率可达100%,检测片段可达1kb,最适为100~500bp。其基本原理基于:当ds DNA沿变性剂浓度梯度增加的聚丙烯酰胺凝胶电泳时,DNA分子中解链温度(Tm)低的部分逐渐变性解链。一般总是碱基错配部分的异源DNA双链Tm最低,总是先解链,导致DNA迁移速度明显下降。因此,当解链的DNA链中有一个碱基改变时,会在不同的时间发生解链,从而因电泳速度变化的程度不同而被分离,达到分离野生型与突变型DNA片段的目的。DGGE的成功有赖于DNA内部低温解链部分变性后迁移率的改变。但在相当于最高Tm的变性剂浓度的位置,相应的DNA片段可能全部解链,使实验无法检出存在于高Tm DNA片段中的碱基替换。为解决此问题,在合成的PCR引物的5′端加一段40~50bp的GC夹,以利于检测发生于高熔点区的突变。在DGGE的基础上,又发展了用温度梯度代替化学变性剂的温度梯度凝胶电泳法(temperature gradient gel electrophoresis,TGGE)。
(4)随机扩增的多态性DNA(random amplified polymorphic DNA,RAPD):是应用随机引物扩增并寻找可作为分子标记的多态性DNA片段。尽管RAPD技术诞生的时间很短,但由于其独特的检测DNA多态性的方式以及快速、简便的特点,使这个技术已渗透于基因组研究的各个方面。RAPD技术建立于PCR技术基础上,它是利用一系列(通常数百个)不同的随机排列碱基顺序的寡聚核苷酸单链(通常为10个核苷酸)为引物,对所研究基因组DNA进行PCR扩增,聚丙烯酰胺或琼脂糖电泳分离,经EB染色或放射性自显影来检测扩增产物DNA片段的多态性,这些扩增产物DNA片段的多态性反映了基因组相应区域的DNA多态性。RAPD所用的一系列引物DNA序列各不相同,但对于任一特异的引物,它同基因组DNA序列有其特异的结合位点,这些特异的结合位点在基因组某些区域内的分布如符合PCR扩增反应的条件,即引物在模板的两条链上有互补位置,且引物3′端相距在一定的长度范围之内,就可扩增出DNA片段。因此,如果基因组在这些区域发生DNA片段插入、缺失或碱基突变就可能导致这些特定结合位点分布发生相应的变化,而使PCR产物增加、缺少或发生相对分子质量的改变。通过对PCR产物检测即可检出基因组DNA的多态性。分析时可用的引物数很大,虽然对每一个引物而言其检测基因组DNA多态性的区域是有限的,但是利用一系列引物则可以使检测区域几乎覆盖整个基因组。因此,RAPD可以对整个基因组DNA进行多态性检测。另外,RAPD片段克隆后可作为RFLP的分子标记进行作图分析。
(5)其他方法:在精细基因图谱制作、基因连锁发现、亲子鉴定及群体遗传学等研究中,RFLP分析已逐渐被VNTR、SNP分析、AluⅠ序列多态性分析所涵盖。除了以上几种多态性的检测方法外,基因芯片、单链构象多态性、PCR-DNA测序、PCR指纹图法、PCR-荧光法、PCR-SSP(序列特异性引物)、PCR-SSO(顺序特异寡核苷酸)和PCR-ASO探针法均可应用于DNA多态性分析。
将以上几种基因结构改变的诊断方法总结于表14-1。
表14-1 基因结构改变的诊断方法

三、基因表达异常的诊断
在基因结构未发生改变的情况下,基因表达的异常也可以引起疾病。通过对RNA进行定量和定性分析,既可用于疾病的诊断,也可用于基因治疗效果的监测。常用的基因表达的诊断方法有以下几种。
1.mRNA相对定量分析
(1)斑点杂交或狭线杂交:将提取的待测RNA通过斑点或狭线点样装置点在尼龙膜上,与标记探针进行杂交,然后比较正常对照与待测样品的斑点光密度,可以确定mRNA表达量的相对高低。同样,通过Northern杂交也可检测RNA表达量的变化。
(2)RT-PCR法:从表达某一特定基因的组织中提取细胞总RNA,经反转录合成cDNA,然后经PCR扩增、电泳,比较条带光密度或测定放射性活度,推算出mRNA的相对含量。也可采用定量PCR仪,直接分析mRNA的相对含量。
2.mRNA绝对含量分析 将待测mRNA反转录成cDNA作为PCR扩增的模板,另外再构建一组与待测cDNA相似,但长度相差50bp的“标准cDNA”,并稀释成已知的不同浓度,然后与待测cDNA一起作为模板,在相同的扩增引物的指导下进行竞争性PCR反应,以32P-dCTP作标记。依次测定竞争性PCR后两组扩增产物的放射性活度,经计算处理后可得出mRNA的绝对含量。
3.mRNA长度变化分析 mRNA长度的变化可通过Northern杂交来观察。也可根据RT-PCR产物电泳条带的位置,判断cDNA的大小,确定相应的mRNA区段是否有长度改变。通过长度分析可以初步确定基因中是否有缺失或插入,以及是否有mRNA剪接加工缺陷的可能,随后进一步进行测序确定。
四、外源DNA的检测
病原体的检测对感染性疾病的防治和流行病学调查具有重要意义,以前对病原体的检测需要应用微生物学、免疫学和血清学方法,灵敏度较低或特异性不高,不能用于早期诊断。如今许多病原体包括细菌、病毒、支原体、衣原体、立克次体和寄生虫的基因结构已被阐明,应用核酸杂交技术和PCR技术即可早期、快速、敏感、特异地确定病原体的存在。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。











