



第一节 遗传性球形红细胞增多症
一、流行病学
遗传性球形红细胞增多症(hereditary spherocytosis,HS)是一组以外周血红细胞呈球形改变为特征的溶血性贫血,在世界各地均有发病,以北欧后裔最常见,发病率大约在1∶(2 000~5 000),我国尚无确切的发病率统计数据。根据上海长海医院对506例贫血黄疸病因分析,在先天性溶血病因中,红细胞膜病约占42%、红细胞酶病占23%、血红蛋白病占35%;在红细胞膜病中,又以HS最多,占84%。据国内文献报道,HS在北方地区居遗传性溶血性贫血首位。
二、分子遗传学
HS的遗传类型很复杂,多数患者(约3/4)为常染色体显性遗传,约1/4为隐性遗传,也有新生突变及非典型的遗传缺陷。从文献病例报道看,隐性遗传HS患者的病情一般比显性遗传患者的严重,但是典型的常染色体显性遗传纯合子很少见,可能因致命的重度贫血而难以生存。已报道的少数纯合型HS病情严重、需依赖输血,其双亲表现轻至中度贫血或无贫血,其中多数为血缘姻亲。即使同一家系中的患者,临床表型的差异也很大,与携带或并存不同的等位基因有关。HS临床表现明显不同的另外一些解释是基因缺陷外显率差异大、存在修饰后的等位基因而影响膜蛋白的表达,或者是由于缺陷的组织特异性镶嵌现象。HS的分子研究表明,HS有较高的自发性基因突变率,这些新生突变多发生在CpG岛(基因组中富含CpG的单拷贝非甲基化基因座),引起缺失或插入,以常染色体隐性遗传方式传给后代,这也使HS的遗传类型和临床表型更为复杂多样。按遗传型和临床表型可将HS细分为杂合子的静止型、轻型和较严重型,纯合子的临床静止型、轻型和重型复合型杂合子。
已经明确的红细胞膜蛋白基因在染色体定位如下;收缩蛋白(谱蛋白,spectrin)的Spα亚基(band 1protein)和Spβ亚基(band 2protein)分别位于1号染色体和14号染色体;锚蛋白(ankyrin,band 2.1protein)定位于8号染色体;带4.2蛋白(band 4.2protein,pallidin)定位于15号染色体;带4.1蛋白(band 4.1protein)定位于1p36.2-p34;带3蛋白(阴离子交换蛋白,anion exchange,AE1,band 3protein)定位于17q21-q22;肌动蛋白(beta-actin)定位于7pter-q22;血型糖蛋白C(glycophorin C,GPC)定位于2q14-q21(表4-1)。
表4-1 红细胞主要膜蛋白的基因定位及相关参数

续 表

早在1975年有研究报告提示,HS有染色体改变,8号、12号交互易位,易位也可发生在6号染色体上HLA位点附近。后续研究表明HS可发生8号染色体近端短臂中间缺失,并且在敲除锚蛋白(ankyrin)的nb/nb小鼠得到确证,以后又在不同家系2例缺失锚蛋白的严重HS患儿查证异常核型8p11.2和8p11.23p21.1。
三、病因学
导致HS的最常见原因是红细胞膜骨架与脂质双层膜垂直连接(亦称纵向连接)中的膜蛋白发生病变(“垂直连接”详见第一章),使膜蛋白含量减少或完全缺失。其他病因可以是生化代谢异常,如磷酸化-脱磷酸化异常或阳离子泵异常,也可以是分子内或分子间缔合异常。
红细胞膜垂直连接的关键组分是谱蛋白(spectrin),也称其为垂直连接的中心膜蛋白,它具有ankyrin和protein 4.1的结合位点。βspectrin与αspectrin可以形成异源二聚体、四聚体,作为膜骨架网络主体成分,水平连接(亦称横向连接)构成二维膜骨架网络,通过与跨膜的内在蛋白相互作用即垂直连接(Sp-2.1-band 3、Sp-4.1-GPs以及Sp-4.1-band 3和Sp-2.1-4.2-band 3等),将膜骨架网络附着在双层脂质膜上,使红细胞具有特殊的双面凹圆盘结构和柔韧适宜的变形能力。HS可以由spectrin本身缺陷引起,也可以因为与spectrin垂直连接相关的膜蛋白(如ankyrin、band 3、band 4.1和band 4.2)发生病变,导致spectrin含量或功能缺陷,最后共同通路(final common pathway)都是红细胞球形变化(红细胞膜蛋白垂直连接模式图见图4-1,膜蛋白命名、定位、功能、连接详见第一章红细胞代谢)。
国外已报道的涉及垂直连接的红细胞膜缺陷蛋白有收缩蛋白α-亚基与β-亚基、锚蛋白、带3蛋白和带4.2蛋白。欧美人群中,ankyrin突变引起的常染色体显性和隐性HS最多,占40%~50%,α-spectrin突变引起的常染色体隐性遗传HS和β-spectrin突变引起的常染色体显性遗传HS均占HS的10%,band 3突变引起的常染色体显性HS约占20%,band 4.2蛋白突变引起的常染色体隐性HS相对较少见,但是在日本band 4.2突变是HS最常见病因。
国内文献显示,中国人HS中除了有上述膜蛋白的缺陷以外,还常见带4.1蛋白(band 4.1protein)缺乏,而在欧美人群中band 4.1缺陷主要见于遗传性椭圆形红细胞增多症(HE)。在HS膜蛋白缺陷排序上,中国人群中依次以band 4.1、spectrin和band 3缺陷多见,不同于欧美人群(ankyrin、spectrin和band 3)和日本人群(band 4.2)。
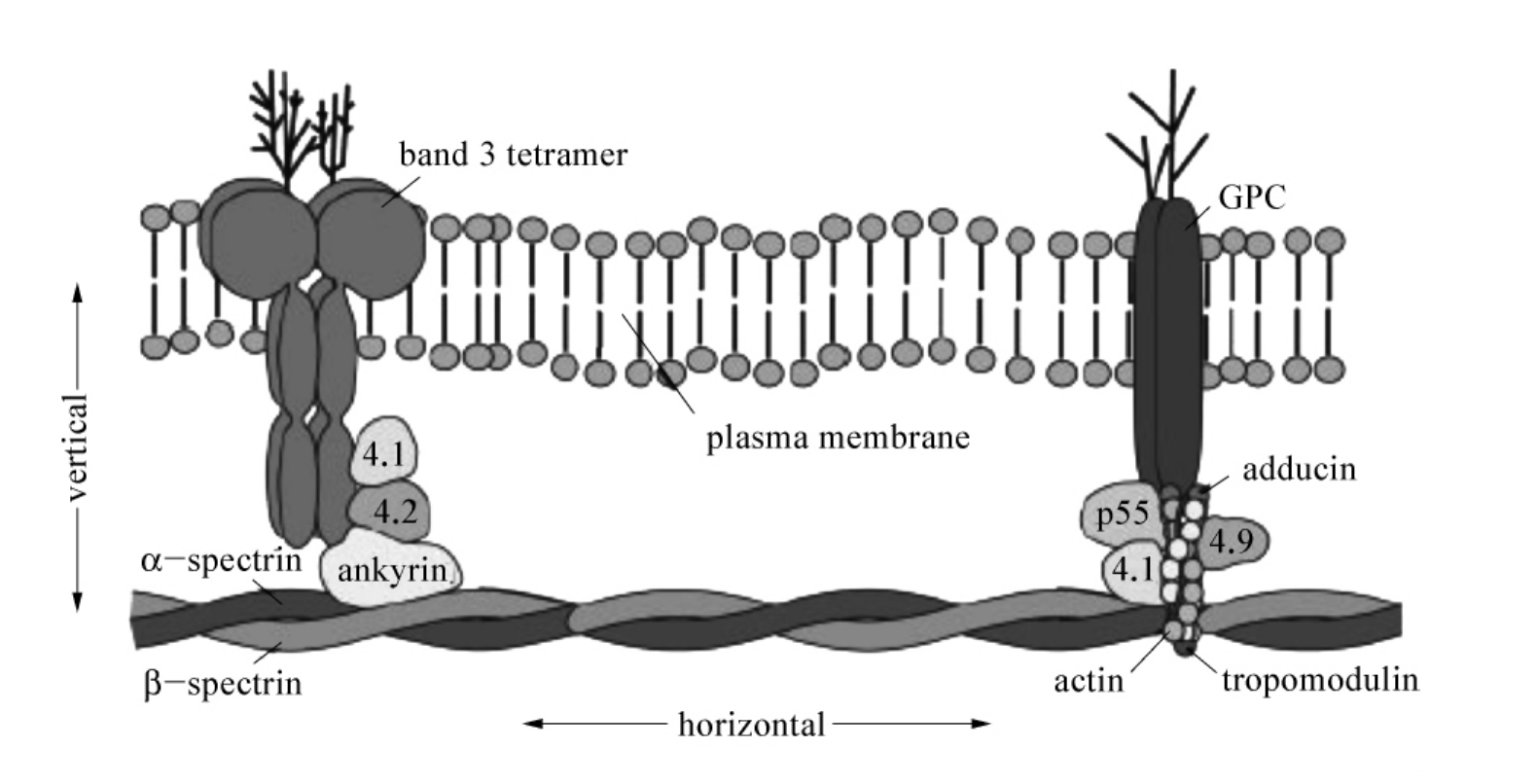
图4-1 红细胞膜蛋白纵向连接与横向连接模式图
β-spectrin:收缩蛋白(谱蛋白)β亚基(band 2protein),垂直连接关键组分;αspectrin:收缩蛋白(谱蛋白)α亚基(band 1protein);ankyrin:锚蛋白(band 2.1protein);band 3:带3蛋白(阴离子交换蛋白,band 3protein,AE1);4.1:带4.1蛋白(band 4.1protein);4.2:带4.2蛋白(band 4.2protein);4.9:带4.9蛋白(band 4.9protein,Dematin);p55:p55蛋白(p55protein);actin:肌动蛋白(band 5 protein);glycohporin C/D:血型糖蛋白C/D(GPC/GPD);adducin:加合蛋白;tropomodulin:原肌球调节蛋白;plasma membrane:脂质双层膜。
引自Birkenmeier CS and E Barker JE.J Pathol,2004,204:450~459
四、分子病变
迄今为止的研究表明,HS的分子缺陷是极不均一的,可由多种膜蛋白异常引起,许多突变是独特的,未在其他家系重现,亦即每个家系都存有引起球形红细胞增多症的独特基因,没有一种分子突变具有共性,而在血红蛋白病和红细胞酶病研究中,均鉴定出特定人种、地区高频概率的突变热点。
已鉴定的HS病变膜蛋白基因序列中多数有点突变,还有mRNA加工异常、基因缺失或低表达等突变体。在显性HS中,常见ankyrin、band 3和β-spectrin无义突变和移码突变。在隐性HS中,常见ankyrin、α-spectrin或band 4.2双杂合型缺陷。
(一)收缩蛋白(谱蛋白,spectrin)缺陷
1.α-收缩蛋白缺陷 隐性遗传的HS常见α-Sp缺陷,多为重度溶血。α-肽链第九重复片段中969位Ala→Asp的点突变,可引起正常α-Sp合成减少或分子不稳定。由于在正常红系细胞中α-Sp的合成量远远超过β-Sp合成量,为β-Sp的3~4倍,所以在一个α-Sp等位基因失活(多为αLEPRA,遗传自正常个体的沉默基因),而另一个等位基因正常的个体,α-Sp的产生仍大于β-Sp,因此仍有正常量的α-Sp和β-Sp结合而成的spectrin异二聚体被装配在膜上,该个体可以成为无临床症状的分子缺陷的HS携带者。然而,如果是α-Sp分子缺陷的纯合子或双杂合子,则可表现出α-Sp合成量明显降低,装配在膜上的Sp也显著减少。例如具有不同α-Sp基因缺陷的双杂合子αLEPRA/αPRAGUE,其中一个等位基因αLEPRA为上游内含子剪接突变,使得α-Sp转录本的合成量低于正常6倍。在双杂合子αLEPRA/αBug Hill,其中αBug Hill发生αⅡ区域的氨基酸置换。这些合并αLEPRA等位基因的双杂合子都表现为α-spectrin明显缺乏的重度HS。
2.β-收缩蛋白缺陷 显性遗传的HS可见β-Sp缺陷。β-肽链中第202位的Trp→Arg点突变(β-spectrinKissimmee),可导致β-Sp合成减少或不稳定,由于突变位点正好位于β-Sp与带4.1蛋白结合的高度保守区,所以这种点突变的β-Sp就失去了与带4.1蛋白的结合功能,也因此影响与肌动蛋白的连接。有些家系除了点突变(β-spectrinHouston)还存在移码突变,可能减少β-spectrin mRNA累积。
(二)锚蛋白(ankyrin,band 2.1protein)缺陷
在显性遗传HS和隐性遗传HS均可见锚蛋白缺陷,基因突变多发生在锚蛋白编码区,也有突变发生在启动子上,使锚蛋白表达量减少。在隐性遗传HS,可见双杂合型突变,既有锚蛋白的启动子或5′非编码区异常,又合并错义突变。同收缩蛋白变异一样,锚蛋白突变位点在各个HS家系均不同,20%为新生突变。锚蛋白通过Sp-2.1-band 3连接而成为收缩蛋白垂直连接于膜上的主要位点,所以红细胞膜锚蛋白缺乏可直接影响Sp-2.1的连接,此时尽管患者Sp的mRNA含量正常,但由于锚蛋白减少,所连接的Sp亦减少。所以,患者可表现为锚蛋白与收缩蛋白同步缺乏,锚蛋白为原发缺陷,收缩蛋白为继发缺陷。
有些病例中发现8号染色体锚蛋白基因位点发生缺失或易位,这种缺失可能产生相邻基因综合征(contiguous gene syndrome),除了表现为球形红细胞增多症以外,还伴发智力发育迟缓、特殊面容及性腺发育不全。
(三)带3蛋白(band 3protein,anion exchanger,AE1)缺陷
约有20%显性遗传的HS有带3蛋白的缺失,绝大多数为杂合子,患者临床多表型为轻至中度的溶血,带3蛋白缺陷杂合谱蛋白缺陷的双显性个体可以导致新生儿死亡或严重的溶血性贫血。带3蛋白基因缺陷的纯合子较少见,已报道的葡萄牙人band 3Coimbra(Y488M)纯合子出现新生儿水肿,意大利人band 3Neapolis(16+2T→C)为剪接缺陷抑制翻译起始,也导致严重贫血,危及生命,而纯合子的双亲均为轻型HS。
带3蛋白缺陷HS患者的红细胞可呈现蘑菇样(又称乒乓球拍样)或者“鳌钳”(pincered)样特殊形态学改变(见后面图4-6),很多患者还同时伴有带4.2蛋白缺乏,与带3蛋白突变后影响与带4.2蛋白结合有关。目前已鉴定出50余种带3蛋白突变型,突变遍及带3蛋白的各个分区(胞质区、穿膜区和膜外侧区,详见第一章红细胞代谢),主要见于胞质区和穿膜区。许多点突变导致穿膜区域的带3蛋白保守肽段上精氨酸被替换。这些精氨酸均位于跨膜螺旋结构近胞质区末端,可作为终止穿膜信号,有助于维持跨膜片段的精确定位和朝向。发生突变后,可能影响band 3合成后的折叠和插入内质网、最终组装进入红细胞膜的过程。带3蛋白基因突变可导致基因表达量减少,表达产物在红细胞膜中含量也减少。但是,也有一些突变并不改变基因表达量,例如在带3基因818密码子后插入10个核苷酸可导致移码突变,突变体band 3mRNA水平是正常的,但是带3蛋白含量比正常少25%~40%,Sp/band 3之比增加,原因可能是异常带3蛋白分子不稳定,以及从细胞中释出增加。这种带3蛋白分子与带2.1蛋白的结合及其自身在膜中的装配与聚合均有缺陷。带3蛋白分子在膜中聚合成四聚体才能与带2.1蛋白结合,带3蛋白突变后影响二聚体与四聚体转换,而二聚体不能与带2.1蛋白结合,于是病变的带3蛋白可随同膜脂微囊泡一起脱落而丢失,导致带3含量减少。
(四)带4.2蛋白(band 4.2protein,pallidin)缺陷
遗传性band 4.2缺乏HS呈隐性遗传,在日本人群中相对较为常见。带4.2蛋白通过多种方式稳定膜骨架纵向和横向的连接:通过与带3蛋白结合,稳定锚蛋白和带3蛋白之间的连接;通过与带4.1蛋白结合,形成细胞骨架中结合复合物;带4.2蛋白还可以促进SpDSpD之间的连接。带4.2蛋白的基因发生点突变(如142Ala→Thr点突变)或移码突变,可引起膜骨架与膜的结合削弱,使膜丢失,红细胞呈球形改变,凝胶电泳显示带4.2蛋白含量缺乏,在纯合子可完全缺失。带4.2蛋白缺乏的患者溶血程度多为轻至中度,有报道红细胞形态除了球形变还存在口形、卵圆形或棘形红细胞等。
继发性带4.2蛋白缺乏见于带3蛋白突变,带3蛋白胞质区与带4.2蛋白的结合位点中327Pro→Arg点突变,可导致带4.2蛋白与带3蛋白结合减少,因而丢失带4.2蛋白。
继发性带4.2蛋白缺乏还可见于胆道梗阻病人。有研究显示,胆道梗阻黄胆病人红细胞膜带4.2蛋白减少或完全缺失,红细胞呈靶形(target)或马刺状(spur),这种红细胞膜还有脂类含量异常,尤其是胆固醇和磷脂酰胆碱含量升高,在手术去除梗阻后带4.2蛋白的含量可恢复正常。
(五)带4.1蛋白(band 4.1protein,band 4.1R)缺陷
带4.1蛋白是细胞骨架与膜脂质双层相连的重要纵向连接点,同时也是膜骨架网络横向连接三联物(Sp-actin-band 4.1)的主体成分。band 4.1的酶解肽段一区含有GPC、GPD、band 3结合位点以及与膜脂双层中磷脂酰丝氨酸的结合位点,三区含有spectrin和F-actin的结合位点,主要通过Sp-4.1-GPs和Sp-4.1-band 3等复合物连接方式参与膜蛋白与脂膜的纵向连接。
国外文献报道带4.1蛋白含量缺乏主要见于遗传性椭圆形红细胞增多症(HE)。在部分HS,出现带4.1蛋白功能障碍,即带4.1蛋白与谱蛋白等骨架蛋白交联缺陷。
国内文献显示带4.1蛋白缺失是中国人群HS常见原因之一,部分家系有明确溶血家族史。在形态学上,带4.1蛋白缺陷可引起红细胞球形变化和球口形变化,也可以引起红细胞筛孔状(screenmes)变化(图4-2)。筛孔状红细胞在光镜和相差镜下均显示有“咬痕”(bite cell),膜表面似筛网,在电镜下观察可见细胞上有数量不等、大小不一、深浅不均的凹陷,band 4.1a亚基完全缺失。在HS,带4.1蛋白缺陷还可表现为band 4.1a亚基缺失、a/b亚基比值倒置、总量减少及伴有异常区带出现(图4-3)。

图4-2 筛孔状(screenmes)红细胞形态扫描电镜图像
A:×3 000;B:×10 000
五、溶血机制
HS红细胞最主要的溶血原因是膜表面积/体积之比下降、膜变形能力减弱、渗透脆性增加。最主要的溶血场所在脾脏,与脾脏结构相关。
(一)红细胞膜表面积缺失机制
1.膜表面积绝对缺失 红细胞膜蛋白垂直连接中的成分缺失将导致膜表面积绝对减少,膜骨架蛋白spectrin、ankyrin、band 4.2、band 4.1和嵌膜蛋白band 3是文献报道最多的HS缺陷膜蛋白。
HS红细胞膜表面积丢失的途径至少有3种假说:①单独spectrin缺失或spectrin合并ankyrin缺失,使脂质双层膜在内侧面与膜骨架连接缺陷。在正常红细胞,膜骨架网络形成几近单分子的亚层膜,衬于脂质双层膜内层面,覆盖面积占膜内表面积的1/2以上。spectrin为主体的膜骨架蛋白缺乏将导致膜骨架网络密度减小,垂直连接减弱,脂质双层膜与骨架解偶联,带3蛋白在脂质双层膜中的侧向运动增加,结果,不能被骨架直接支持的“无支点”的脂质区域形成脂质微泡,在红细胞循环中可自发丢失,使膜表面积逐渐减少,变形能力减弱。丢失的微泡直径为0.2~0.5μm,可含有band 3,不含spectrin。②band 3分子缺陷削弱了其与骨架蛋白连接,由于band 3穿膜区域多次跨越脂质双层膜,可能作为界面脂质的实质成分随同其边界脂质一起形成脂质微泡脱落丢失,致使膜表面积缺失。③band 3分子完全缺失,膜上形成无band 3的脂质双层膜区,可“生芽”形成泡状膜突起,随后以膜微泡从细胞中释出,致使膜表面积减少,细胞呈球形变化。这一假说是基于实验观察,在血影(ghost,除去红细胞内容物后的重封质膜)中,以band 3为主要成分构成的膜内颗粒聚集,可导致膜脂气泡从颗粒缺失区域以微泡的形式脱落。另外,从band 3基因敲除小鼠模型和人、奶牛、斑马鱼等band 3完全缺失红细胞研究观察,也支持这一假说(图4-4)。
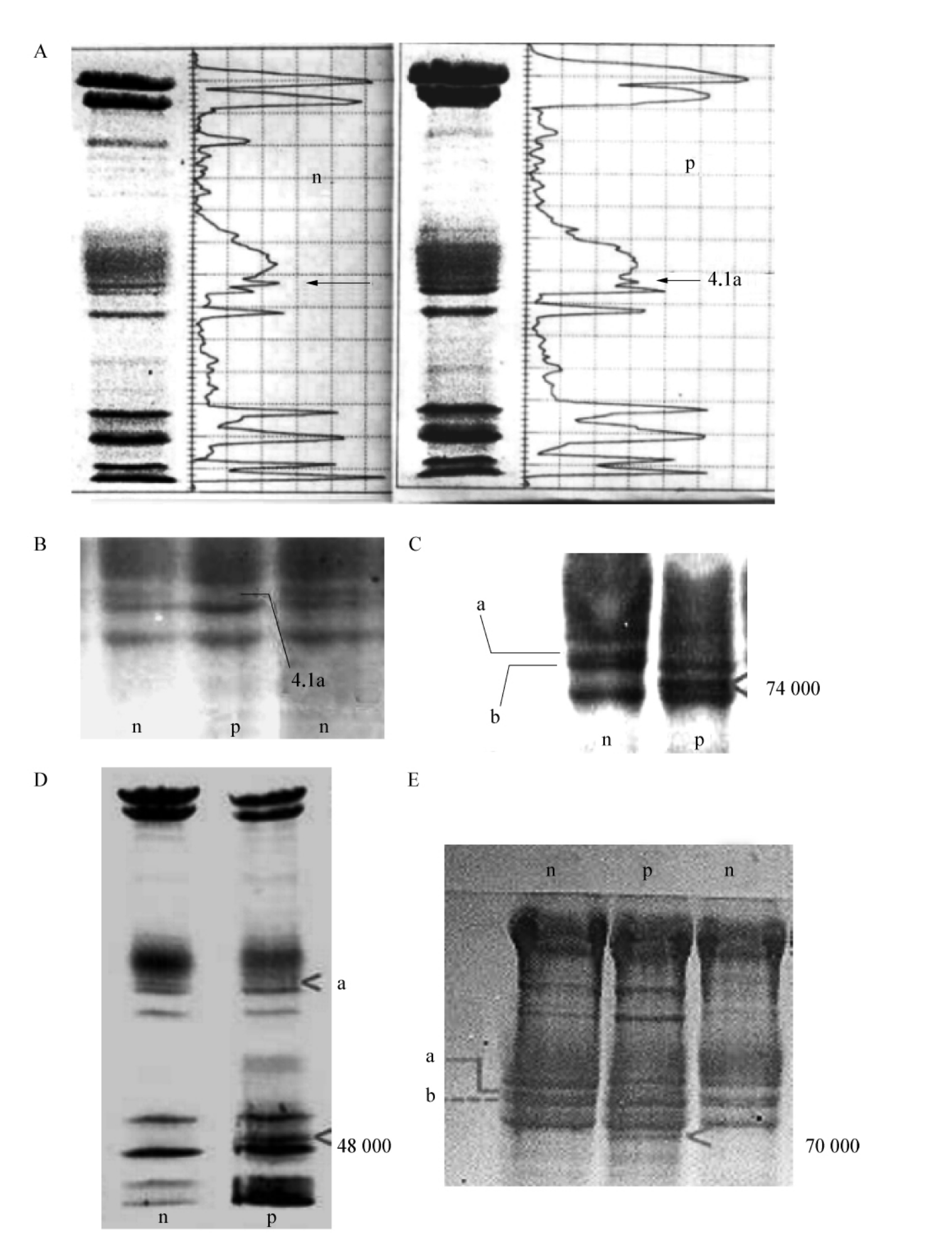
图4-3 中国人红细胞膜4.1蛋白电泳变异型
A:band 4.1a缺乏导致4.1a/b比值倒置;B:band 4.1a亚基完全缺失;C:band 4.1a缺失伴相对分子质量74 000异常蛋白区带;D:band 4.1a部分缺乏伴相对分子质量48 000异常区带;E:band 4.1总量缺乏伴相对分子质量70 000异常区带;n:正常对照;p:HS患者

图4-4 HS红细胞球形变化过程
2.膜表面积相对缺失 HS红细胞由于内侧膜缺陷,使Na+-K+-ATP酶或Ca2+-Mg2+-ATP酶等阳离子泵功能失常,细胞内Na+、Ca2+蓄积,水潴留,细胞内容积增加,可致膜表面积相对缺乏。
红细胞膜表面积绝对缺乏和相对缺乏均使HS红细胞的膜表面积/体积之比下降,形态发生球形、杯形、碗形等改变(见后面图4-6),膜变形功能明显减弱。HS红细胞由于内在不稳定因素,在体外多种条件下包括ATP耗竭、细胞受到切变应力等,也会释出脂质微泡,细胞膜表面积进一步减少,在体外放置可观察到抗凝血样本溶血现象。室温或37℃孵育时,显示出细胞渗透脆性明显增高。
(二)脾脏在HS红细胞溶血中的作用
HS球形细胞选择性地在脾脏破坏,一方面是因为球形细胞变形功能损伤;另一方面是由于脾脏血管具有微循环过滤器样的独特解剖结构。
正常红细胞的表面积(140μm2)与容积(90fl或约85μm3)的比值约为1.5,即实际表面积远大于与容积相称的面积。HS红细胞膜缺失使膜表面积/体积之比下降,没有足够的或称额外多的膜表面积供其变形,以通过脾脏狭窄的微细血管、血窦,所以一旦被脾脏俘获,HS红细胞会进一步受到损伤。
正常时脾脏选择性分拣、扣留代谢减弱和变形性差的衰老红细胞、病变红细胞及带有抗体的红细胞(见第一章第三节)。脾脏截留球变红细胞的主要位点在脾窦壁开口处,在此处从红髓脾索而来的血液进入静脉循环。在大鼠脾脏脾窦口的长和宽分别为2~3μm和0.2~0.5μm,小于红细胞直径的1/2。在人的脾脏,脾窦内孔径最细处≤0.5μm,而红细胞平均体积为7μm。脾脏电镜显示,HS红细胞难以通过这些狭缝,结果不能变形运动的球形细胞蓄积在脾脏红髓,使红髓明显充盈肿大,成为HS脾脏肿大的病理基础和血管外溶血的诊断依据。
脾脏不仅血管微细,易截留吞噬HS细胞,而且脾脏微环境可使HS红细胞膜进一步受到蓄积损伤。脾脏低pH、低糖、低水平ATP、低氧环境以及脾脏吞噬细胞产生的局部高浓度的毒性自由基均增加HS病态红细胞代谢强度,促使细胞膜以囊泡形式(含膜脂、膜蛋白)进一步丢失,细胞密度进一步增加。HS红细胞在脾索平均滞留时间在10~100min,进入脾脏的血液有1%~10%滞留在充血的脾索,而90%的血流快速地进入静脉循环。从脾脏搜集的HS红细胞有脱水现象,推测膜缺陷可能激活了选择性失钾、失水的通路。那些得以逃逸返回循环血液的HS细胞呈现为深染小球形红细胞,在渗透脆性曲线上表现出“拖尾”,这提示存在膜表面积显著减少的细胞亚群,脾切除术后,这一亚群即消失。
大多数HS红细胞最终在脾脏破坏,但也可以在脾外组织器官破坏。HS红细胞膜表面积改变可以促发组织巨噬细胞吞噬,其机制尚不明了,一种可能的途径是脂质双层膜的磷脂分布改变,导致正常时主要分布于膜内侧面的磷脂酰丝氨酸(PS)暴露于脂膜外,促使红细胞黏附巨噬细胞而被其吞噬。研究显示重型HS患者膜磷脂分布确有改变,但是多数HS患者红细胞膜磷脂分布是正常的。
六、临床表现
(一)典型症状
典型的遗传性球形红细胞增多症患者(HS)的特征是有溶血病史和(或)家族史,临床表现黄疸、贫血、脾脏肿大,外周血涂片中红细胞形态以球形变化为主,红细胞渗透脆性增高、网织红细胞计数和网织红细胞生成指数升高,直接抗人球蛋白试验(DAT,Coomb’s Test)阴性。
(二)临床分型
由于前述HS涉及的红细胞膜蛋白变异种类和遗传方式复杂多样,导致HS患者的临床表现差异很大,根据临床表现,可将HS分为轻度HS、无症状携带者、典型HS和重度HS。一般而言,隐性遗传HS症状较严重,而占HS大多数的显性遗传患者通常表现轻至中度溶血症状。虽然红细胞膜遗传缺陷出生时即存在,但是发病年龄可以迟早不一,婴幼儿时期发病者多表现为中重度溶血,成年后发病者多表现为轻至中度临床症状。
1.轻度HS和无症状HS携带者 无实验室溶血证据或溶血程度轻微,网织红细胞计数正常或仅轻度增高,骨髓红系增生可完全代偿而无临床贫血症状;红细胞形态可呈现中空淡染区缩小,但小球形变化不典型;渗透脆性试验可以阳性,也可以阴性;胆红素不增高或轻微增高。这些无症状者受到感染特别是病毒感染和应激条件诱导可使溶血发作,甚至产生溶血危象或再障危象,如有些患者特别是婴幼儿患者首诊原因是传染性单核细胞增多症、微小病毒(parvovirus)感染或流感,还有些患者是因为妊娠或老龄期骨髓对溶血的代偿性增生能力减弱而被确诊。长时间剧烈运动、重体力劳动也可促发溶血,可能活动量增加使脾脏血流量增加、脾脏破坏HS红细胞增多所致。
2.典型HS 即中度HS,早期轻度黄疸可能是唯一症状,大多数患者呈慢性溶血过程伴间歇性急性发作,慢性期主要为血管外溶血,表现为未结合胆红素增高、皮肤巩膜黄染、脾脏渐进肿大,轻至中度贫血、网织红细胞计数增高。骨髓增生代偿良好时可能无贫血,但是患者网织红细胞生成指数(RI)通常大于2。半数以上病例有HS阳性家族史和新生儿溶血病史。
3.重度HS 多见于隐性遗传HS,血红蛋白可低至40~60g/L如收缩蛋白缺乏的隐性遗传HS。在婴幼儿期可发生危及生命的溶血,需依赖输血或换血生存。如果父母双方均有红细胞膜蛋白显性缺陷,则获得双亲遗传缺陷的胎儿可能发生胎儿水肿和死胎。
欧美学者根据患者血红蛋白等指标提出HS临床分型、诊断提示及脾切除术指征,见表4-2。
表4-2 HS临床分型与切脾指征

引自Brit J Haematol.2004,126:455;Seminars in Hematology,2004,41:118~141
(三)并发症
1.胆石症、胆囊炎 发生率高,胆结石总发生率占HS的50%,尽管有些患者溶血性贫血程度很轻,也会发生胆结石。两岁以内婴儿很少见胆结石,但是随着年龄增长,胆结石发生率明显增加,据国外数据统计,2~10岁患儿约为5%,11~50岁患者约为45%,老年患者约为65%。HS患者同时合并其他红细胞遗传缺陷(血红蛋白病或红细胞酶病)或合并遗传性肝细胞性黄疸(吉尔伯特综合征等)会加重溶血、黄疸,增加胆石症的风险。
2.贫血危象
(1)溶血危象:较少见,仅在某些感染时发生,如传染性单核细胞增多症,由于单核巨噬细胞系统(旧称网状内皮系统)吞噬功能增强导致病变红细胞破坏增多而加重溶血、贫血,多呈自限性。
(2)再障危象:较少见,症状重,危及生命。主要病因为病毒感染尤其是细小病毒B19感染,征象为骨髓红系增生抑制、流感样症状和传染性红斑(第五病)症状,出现发热、寒战、嗜睡、抑郁、恶心、呕吐、腹痛有时伴有腹泻、呼吸道症状、肌肉关节疼痛,面部、躯干和四肢出现红色斑丘疹。该病毒特异感染红系前体细胞,抑制其发育成熟,导致贫血,还可伴发中性粒细胞减少和血小板减少。孕妇感染细小病毒极其危险,可致胎儿贫血、水肿甚至死亡。
(3)巨幼细胞性危象:在溶血的基础上发生巨幼细胞性贫血,加重贫血程度。缺乏叶酸引起的巨细胞性贫血极少见,典型的缺乏病例见于叶酸需求量增加的患者如再障危象恢复期患者、孕妇、儿童及老年患者。曾有报道孕妇巨细胞贫血危象为HS首诊症状。
3.与膜蛋白有关的HS非红系症状 大多数HS病例的临床症状集中于红系表现,可能由于非红系的膜蛋白类似物与红系膜蛋白分属不同的基因编码,或者是由于非红系膜蛋白有组织特异性选择剪接。然而,在有些HS家族中已发现和报道并存神经肌肉系统的异常,如脊髓退行性变、心肌病或智力发育迟缓。HS并发精神运动性阻滞(psychomotor retardation)可出现眼部血管样条纹症,有8号染色体异常,而ankyrin的基因也定位于8号染色体。研究发现红系ankyrin和β-spectrin在肌肉、脑特别是小脑也有表达,HS患者分布于这些部位的膜蛋白可能也有缺陷。这一推测在研究ankyrin的突变的HS动物模型nb/nb小鼠中得到进一步证实,这种小鼠的小脑浦肯野细胞(Purkinje cells)中ankyrin的变性和缺失,可伴随HS发生神经系统症状。
没有HS的个体可发生远侧肾小管酸中毒(distal renal tubular acidosis,dRTA),这是一种band 3家族蛋白突变导致离子转运异常的肾脏遗传病。绝大多数band 3突变HS的杂合子患者具有正常的肾脏酸化功能,有两个家族例外,他们均为band 3mRNA加工突变(band 3Pribram、band 3Campinas),同时遗传HS和肾脏酸化功能缺陷,肾小管性酸中毒的发病机制尚不明确。

图4-5 HS患者内踝皮肤变化
4.其他并发症 严重HS可致患儿生长缓慢、性成熟延迟,出现髓外造血灶和类似地中海贫血容貌。HS患者反复输血或者具有两种遗传缺陷如HS携带血色病基因,也可以继发血色病。
其他相对少见的并发症还包括痛风、慢性皮炎和下肢溃疡,见于成年患者。皮肤病变易发生在近踝关节处,溃疡较少见,但常见内外踝部位皮肤粗糙、发黯(图4-5)。痛风和下肢合并症在脾脏切除术后通常消失,但是胸部、脊椎旁等处扁骨端髓外造血灶不会消失而是发生脂肪化改变,在胸片上一直能显现,易误诊为赘生物,须加以鉴别。
有十余例报道HS同时发生恶性血液病包括骨髓增生异常综合征和白血病,究竟恶性血液病是随机发生还是因为长期造血应激增强所致尚无定论。
七、诊断与鉴别诊断
临床诊断可确定溶血存在的一般证据,实验室溶血项目检查才能提供HS确诊依据。最主要的HS诊断参数见表4-3。
表4-3 最主要的HS诊断参数

(一)临床诊断
1.家系与病史 显性遗传HS有家族史,表现轻至中度溶血症状。无家族史的患者通常贫血程度较重。部分患者有新生儿黄疸病史,有感染、感冒、劳累引发黄疸贫血或加重溶血病情的病史。
2.临床指征 典型的HS见皮肤巩膜黄染、不同程度贫血、脾脏肿大等溶血性疾病通症,可伴有胆结石、胆囊炎,成年患者可出现痛风、下肢溃疡等症状。部分患者可见到副脾和髓外造血灶。轻型HS可无贫血,黄疸轻微,也可表现无贫血而黄疸明显、脾脏有不同程度肿大。
3.其他症状提示 某些感染尤其是细小病毒感染后发生溶血、孕妇不明原因溶血、青少年不明原因黄疸脾大、胆石症等情况应做HS排查实验。
(二)实验室诊断
1.具备溶血证据 典型HS患者存在红细胞加速破坏证据,如未结合胆红素增高、LDH增高、结合珠蛋白减少,以及红系代偿增生如网织红细胞计数增高。轻型HS的这些数据变化不明显,但是可有网织红细胞生成指数(RI)增高(通常≥2),有诊断提示意义。
2.血象提示 MCHC增高有诊断意义,反映出HS红细胞有表面积减少、细胞脱水、厚度增加。病程早期MCV可有较明显降低。单项指标MCHC对HS诊断的敏感性约为70%、特异性为86%。网织红细胞计数明显升高、频繁输血、缺乏叶酸等因素可部分抵消或完全掩盖MCHC和MCV的变化。
3.外周红细胞球形形态和浓染等异形变化 典型HS患者的红细胞见明显球形变化,即中心淡染区缩小(正常红细胞中空区域直径约为完整细胞直径的1/3),平均细胞直径减小(正常红细胞平均直径7.2μm)、厚度增加(正常红细胞盘形边缘厚度2.5μm),显现出体积缩小、血红蛋白密度增大,浓染,以至于有些球变细胞无中空区。在重度HS这种体积减小的浓染细胞明显增多,大小不均,形态异常。戊二醛固定后镜下观察三维形态,可见到明显的球口形细胞,还可出现中空区偏位的杯形、碗形等细胞,均为球形变化过渡中的异形红细胞。其他异形红细胞还可见蘑菇形(乒乓球拍形)或螯钳形、锯齿状或棘刺样、拖尾、不规则异形。在一个HS个体,可以见到上面描述的多种异形红细胞,而某些形态在特定类型患者多见,有诊断提示意义,如在band 3缺陷HS更易见到蘑菇形红细胞,脾切除后蘑菇形细胞可消失;而band 4.2缺陷多见口形样球变细胞;合并缺失spectrin和ankyrin的HS易见不规则异形及棘状球形红细胞(图4-6)。

图4-6 HS常见红细胞形态学变化
A:正常红细胞形态,中空区约为细胞直径的1/3,10×100;B:HS携带者,体积正常,中空区缩小,部分球变,10×100;C:大小不均,小球形变,见于β-spectrin缺乏等各类HS,10×100;D:明显小球形变,见于band 4.1缺乏等各类HS,10×40;E:口形样球形变,多见于band 4.2缺乏HS,10×100;F:深染红细胞为主HS,10×100;G:深染红细胞,箭头示碗形红细胞,10×100;H:箭头示杯形红细胞,10×100;I:棘形(锯齿形)红细胞,深染为主,多见于合并ankyrin/spectrin或两种以上膜蛋白缺陷的HS,10×100;J~L:蘑菇形(螯钳形、乒乓球拍形),易见于band 3缺乏HS,10×100
在轻型HS患者,由于膜表面积丢失轻微,其红细胞球形变化不甚明显,而是表现为细胞厚度有所增加的“肥硕”的盘形(“fat”disk)。
一般认为球形红细胞大于5%可以诊断HS,但是由于HS红细胞可形成多种形态的膜表面积减小的异形细胞,所以仅强调球形细胞大于5%作为诊断标准是不够准确的。有些患者球形细胞可能少于5%,但其深染细胞和其他异形细胞明显增多。据分析,浓染细胞比小球形细胞对HS鉴别诊断更具有特异性,不过小球形细胞的比例增高与溶血的严重程度相关。
在正常新生儿血涂片上可见到球形红细胞,所以新生儿诊断HS最好6个月后复查。
4.红细胞渗透脆性增高
(1)盐水渗透脆性试验:渗透脆性试验(osmotic fragility test,OF,OFT)是在体外测定悬浮红细胞在低渗溶液中的抗渗透能力即细胞吸水膨胀能力,常用不同梯度浓度的氯化钠溶液进行试验,故又称盐水渗透脆性试验。正常红细胞膜可以自由通透水分子,使体积逐渐膨胀,达到“临界溶血体积”时细胞即溶血破裂,释出血红蛋白等胞内容物。HS红细胞丢失膜组分而使表面积缺失,抗渗透能力下降,所以达到HS细胞临界溶血体积的溶液渗透压明显高于正常红细胞,以此代表球形细胞渗透脆性增高。

图4-7 HS渗透脆性曲线特征
灰度:渗透脆性正常范围;typical:典型HS渗透脆性曲线;diagonal:重度HS渗透脆性曲线;tailed:拖尾渗透脆性曲线;lysis:溶血百分率
引自Dacie J.Hereditary spherocytosis(HS).In The Haemolytic Anaemias,3rd ed,vol 1.Edinburgh,Churchill Livingstone,1985,134~215
根据相应渗透压值的溶血百分比作图,得到渗透脆性曲线,HS明显曲线右移。若出现“拖尾”曲线,提示存在经脾处理后膜表面积显著减少的细胞亚群,切脾后该拖尾消失(图4-7)。
(2)孵育后渗透脆性试验:在欧洲该试验作为HS诊断金标准。红细胞抗渗透能力主要决定于红细胞膜表面积和体积之比,HS红细胞有膜的缺失,同时也存在细胞脱水,脱水严重的球形细胞由于体积缩小而使得渗透脆性试验不敏感。在没有明显球形变化的轻型HS,渗透脆性变化也不明显。此时可应用孵育后渗透脆性试验,在37℃条件下,孵育24h,因为HS红细胞膜存在渗漏和不稳定性,较正常红细胞更容易失去膜表面积,在孵育过程中球形细胞进一步丢失膜组分,可以得到阳性结果,提高检出率,但同时该实验特异性下降。
有20%的轻症患者对OFT或孵育后OFT不敏感而漏诊,可以选择下述其他渗透脆性测定试验。
(3)其他渗透脆性相关试验
1)酸化甘油溶血试验(acidify glycerollysis test,AGLT50):敏感性高于盐水渗透脆性试验,由于使用甘油来延缓细胞在低渗液中膨胀破裂,故得以记录溶血50%的速率时间,该实验用血微量,适用于婴幼儿和重度贫血HS,有些无临床症状的HS携带者也可显示阳性结果。出现阳性结果的疾病还可见于免疫性溶血性贫血、丙酮酸激酶缺陷症、严重的葡萄糖-6-磷酸脱氢酶缺乏症、遗传性胎儿血红蛋白持续增高症、骨髓增生异常综合征、部分透析的肾衰患者、部分孕妇。
2)红细胞双相渗透抗性试验:原理同AGLT50,借助专用仪器,可以定量测定抵抗高渗和低渗溶液的溶血率,对轻度HS敏感性高,用血微量。
3)高渗冷溶试验(hypertonic cryohemolysis test):利用温度差对细胞膜通透性的影响,测定渗透溶血率,有些OFT和AGLT50阴性结果的HS该项测定可显示阳性结果。Ⅱ型先天性红细胞生成异常性贫血(CDAⅡ,HEMPAS)和东南亚卵圆细胞增多症(SAO)也可呈现阳性结果。
4)红细胞自溶血试验:检测红细胞在无菌、无葡萄糖条件下自发溶血程度,敏感性不如孵育后渗透脆性试验,已较少用。
5.红细胞膜蛋白分子缺陷检测 这类检测方法特异性强、敏感性高,但实验条件要求和操作成本亦高,罗列如下,供参考和选用。
(1)电泳检测:约80%的HS患者包括球变明显而OFT阴性的患者可以检出膜蛋白异常,鉴定出在前述HS分子病变机制中描述的各种生物化学表型。血样低渗处理后用十二烷基硫酸钠溶解红细胞膜蛋白,进行聚丙烯酰胺凝胶电泳(SDS-PAGE),染色后灰度密度值扫描定量,计算各膜蛋白相对含量。大多数HS患者红细胞都有一种或多种膜蛋白的含量缺乏或完全缺失(表4-4),有时伴有异常的膜蛋白降解区带。也有使用spectrin/band 3和ankyrin/band 3比值分析spectrin和ankyrin含量变化,但是如果band 3有缺陷或随膜脂微泡丢失,该比值就可能使spectrin和ankyrin测值比实际含量偏高,这类患者切脾后测定比值会下降。
表4-4 常见红细胞膜蛋白缺乏及其原因

(2)放射性免疫标记:比SDS-PAGE敏感性高,用特异结合某一膜蛋白抗体的同位素探针标记,可以精确地定量每个红细胞上各种膜蛋白的拷贝数。该法用于研究膜疾病,因成本高临床少用。
(3)流式细胞仪荧光测定:用伊红-5-马来酰亚胺(eosin-5-maleimide)标记完整红细胞,通过流式细胞仪检测荧光含量,反映Rh相关的整合蛋白和带3蛋白的量,已作为一项新的HS诊断的筛选试验,对HS的敏感性(92.7%)和特异性(99.1%)都非常高,膜上有缺陷则荧光测值明显减低。荧光测值减低还可见于CDAⅡ、SAO、HPP等膜缺陷性疾病,而自身免疫性溶血性贫血该测值正常,可以与遗传性红细胞膜病鉴别。
(4)红系培养细胞分析:用外周血爆式集落形成单位培养出网织红细胞和幼红细胞,研究膜蛋白的合成及其在膜上的组装,仅对重度HS敏感,轻至中度HS不易检出异常,而且操作很烦琐,应用受限。
(5)染色体检查:多数报道阴性,包括国内长海医院检测结果。国外报道ankyrin缺失的个别家系查出8号染色体与12号染色体或6号染色体上HLA位点附近易位,异常核型为8p11.2和8p11.23p21.1。
(6)基因突变分析:可以确定导致膜蛋白缺陷的突变基因,分辨原发缺陷与继发缺陷,分辨隐性遗传或非显性遗传及新生突变。应用多聚酶链式反应(polymerase chain reaction,PCR)的突变筛选技术,包括单链构象多态性(single strand conformation polymorphism,SSCP)、变性高效液相层析法(denaturation high performance liquid chromatography,DHPLC),分析基因组DNA中HS基因的候选区域,进行测序,以分析基因缺陷。应当注意,有些在SDS-PAGE中显示缺陷的膜蛋白,其基因分析结果是正常的,可能原因是继发缺陷,即其他膜蛋白缺失导致该种膜蛋白连接异常而使含量减少,还有可能是该膜蛋白基因有修饰后缺陷或缺陷发生在非编码区。遗传连锁分析(genetic linkage analysis)必须研究大家族样本,不适于新生突变,而HS是新生突变发生率高的遗传病。常见红细胞膜蛋白原发缺陷和继发缺陷及其原因见表4-4。
(三)鉴别诊断
1.球形形态与脆性试验 有多种疾病可以引起球形细胞增多或红细胞中空区域缩小、深染,也可表现渗透脆性增高,例如自身免疫性溶血性贫血(AIHA)、不稳定血红蛋白病(uHb)、红细胞丙酮酸激酶缺陷症(PKD)和葡萄糖-6-磷酸脱氢酶缺陷症(G6PDD)、附红细胞体病(eperythrozoonosis,人畜共患病)等,都有相似的溶血体征,但MCHC增高仅对HS特异。可用抗人球蛋白试验(Coombs’test,柯姆试验)鉴别诊断AIHA,应注意反复输血的HS患者可以出现Coombs阳性反应,以抗C3抗体阳性相对多见,易误诊为IHA。异丙醇试验和Heinz小体生成试验用以鉴别uHb,专项酶活力测定排除红细胞酶病,附红细胞体病则直接镜检观察受感染的血细胞。在形态学改变上,某些遗传性口形细胞增多症和罕见的Rh缺陷综合征可以出现球口形红细胞,而干瘪红细胞增多症、血红蛋白病Hb SC和Hb CC均可见深染红细胞增多,容易与HS混淆,需做家系调查与相关检测加以区别。
大球形红细胞增多症(macrospherocytosis)为一种罕见的Na+、K+通透异常的溶血性疾病,红细胞形态有球形变化趋势但是不典型,故又被称为“非典型HS”,应注意鉴别。这种疾病脾切除术疗效不佳,而且术后血栓发生率高。
其他可出现明显球形红细胞变化的疾病见于新生儿ABO不相容溶血、溶血性输血反应、梭状芽胞杆菌败血症、重度烧伤、急性红细胞氧化性损伤、重度低磷酸盐血症,以及毒蛇、蜘蛛、蜜蜂叮咬引起的溶血。
2.合并症影响 HS同时并存珠蛋白生成障碍性贫血可部分纠正红细胞球形变化,渗透脆性试验初始溶血NaCl浓度无增高变化,完全溶血NaCl溶度可降低。铁缺乏时,也可使HS球形形态和升高的渗透脆性有所改善,但是红细胞寿命仍旧缩短。这两种情况均可在血涂片上观察到并存中空扩大细胞和中空缩小细胞,需做血清铁蛋白和血红蛋白病检测予以甄别。叶酸和维生素B12缺乏可以使HS红细胞MCV增大,减弱HS相关实验的敏感性。并发阻塞性黄疸的HS患者其红细胞球变表现、渗透脆性、溶血程度会有短暂的改善,可能是从异常血浆脂蛋白增加吸收磷脂和胆固醇,使红细胞膜表面积增加之故,球形细胞可转变为盘形细胞,而单纯患阻塞性黄疸病人的红细胞可以发生靶形变化,不存在溶血证据。
3.膜蛋白继发缺陷 膜骨架蛋白结合了某些结构异常的血红蛋白、珠蛋白链或高铁血红素可引起氧化性损伤,导致收缩蛋白等膜蛋白继发性缺陷。所以,应结合其他溶血指标判断膜蛋白定性定量分析结果,排除合并症影响。
4.继发病变 微小病毒B19感染可以诱发HS以及其他遗传性溶血性疾病的慢性溶血过程变为急性发作,也可以导致免疫性溶血性贫血(AIHA)。平时儿童较少见AIHA,与成年人AIHA相比,HS儿童发生病毒性感染引起AIHA的病程一般比较短暂。
5.与肝细胞性黄疸的鉴别 从临床表现、肝功能损伤指标、溶血证据等方面可以区别溶血性黄疸与肝细胞损伤所致黄疸。但是,以非结合胆红素升高为主的遗传性肝细胞性黄疸如Gilbert综合征,与网织红细胞无明显增高的轻型HS有许多相似之处,常难以鉴别,比如有相似的发病年龄、可有家族史、间接胆红素轻度增高、诱发黄疸加重的常见原因相同、苯巴比妥(鲁米那)试验短期可褪黄等。此时,肝细胞组织病理学、饥饿试验以及患者与其双亲的红细胞形态学更具鉴别诊断意义。如果遗传性肝细胞性黄疸与HS并存,可加重溶血症状。
八、治疗与预后
(一)脾脏切除术
1.脾脏切除术疗效 对HS疗效显著。①临床改善:由于形态异常的遗传性球形红细胞主要在脾脏破坏而使其过早地从循环血液中被清除,所以脾脏切除术后,尽管红细胞膜病变仍然存在、病变细胞的寿命仍然是缩短的,但是大多数HS患者的贫血明显改善,黄疸消退,网织红细胞计数几近正常(1%~3%)。有些重症HS在脾切除术后仍有轻度贫血,特别是收缩蛋白含量低于正常对照50%的HS患者,但输血量减少或不再依赖输血治疗。②红细胞形态学特点:脾切除术后,血涂片可见豪-周小体(Howell-Jolly bodies)、棘红细胞、铁粒红细胞(siderocyte),这些细胞原本在脾脏清除,术后存在于循环血液中。患者球形红细胞仍然存在,所以红细胞膜渗透性仍然增高,但是脾脏处理所致膜表面积严重缺失的小球细胞化已不存在,表现为由脾脏截留球形红细胞亚群所产生的渗透曲线“拖尾”现象消失了(图4-7)。
2.脾切除术的指征 HS是否选择脾切除术应从切脾指征和手术风险两方面考虑。推荐行脾切除术的指征:①重度贫血HS患儿,Hb低于80g/L,网织红细胞计数大于10%。②生理活动受影响的中度贫血HS患儿,Hb为80~110g/L,网织红细胞计数8%~10%,存在生长迟缓、骨骼变形、髓外造血、贫血导致重要脏器损伤。③成人HS需考虑贫血程度、脾肿大程度、髓外造血灶的发展情况、胆结石的有无、小腿溃疡并发症等因素。轻度HS(Hb大于110g/L,网织红细胞计数低于8%)可暂不考虑切脾。④手术年龄:考虑到儿童免疫力问题,如需手术,通常在6岁以上实施。近年欧美国家对HS切脾术实施年龄放宽,如果Hb在60~80g/L,网织红细胞计数>10%,可在5岁后切脾;如果Hb<60g/L,可考虑在3岁后切脾(参见表4-2)。
3.手术风险
(1)早期并发症:局部感染、脓毒败血症、出血、胰腺炎。少数患者出现缺血性心脏病,虽发生率低但危险性明显增高。所有HS患者脾切除术前和术后都应注意防止细菌性脓毒症,引发病菌多为耐青霉素的肺炎球菌、脑膜炎球菌、流感(嗜血)杆菌B。在226例成人HS切脾术后调查,败血症发生率约为2%。3~5岁低龄患儿一旦发生术后败血症,病情将非常严重,所以即使是依赖输血的患儿,切脾也不做首选。
(2)远期并发症:①血栓症和肺动脉高压:脾切除术后血象三系均可升高,血小板升高尤其明显。一周至数周内白细胞可恢复正常,而血小板需较长时间逐渐接近正常值,数量过高的血小板可引起肠系膜静脉闭塞或肝门静脉闭塞,所以血小板高于800万×109/L应当服用抗血栓药物以预防栓塞,对于有心血管疾病的中老年HS患者切脾术后要及早用抗血栓药物。有报道在232例40岁以上的HS患者中血栓发生率为13.3%,心肌梗死或中风的发生率为3.4%~15%,其中8人以前施行过血管成形术或冠脉手术。在HS实验小鼠,锚蛋白缺乏组血栓发生率为20%,而α-收缩蛋白缺乏组血栓发生率为100%。②致死性脾切除后感染(overwhelming postsplenectomy infection,OPSI):典型的OPSI是由带荚膜的微生物感染,这是一种少见的但是非常严重的晚期并发症,易发生在术后的最初几年里,术前使用肺炎链球菌疫苗和对术后发热的儿童早期进行抗生素治疗可减少OPSI的发生率。
4.术前术后抗感染处理 发达国家要求所有脾切除患者在术前数周接受多价肺炎球菌疫苗注射,进行免疫预防,儿童必须确保注射脑膜炎球菌和流感(嗜血)杆菌B(haemophilus influenzae B)疫苗;术后使用预防性抗生素如长效青霉素至少5年,并且服用叶酸预防巨幼红细胞贫血危象。国内临床通常在术后短期使用抗生素,低龄儿童视体质和恢复情况选择术后一年内口服或注射长效青霉素,也有在术前和术后使用天然药物来源的免疫调节剂,增强机体抵抗力,有效预防手术感染。目前国内尚无切脾术后感染的调查报告。
5.手术方式 手术方式有脾全切和脾次全切,手术进路有开腹与腹腔镜选择。脾全切对HS疗效显著、确切,但是有可能引起上述并发症。脾次全切手术效果虽然不及脾脏全切,但小样本统计手术并发症有所减少。欧洲一些医院提倡对严重贫血、依赖输血、生长迟缓的婴儿或低龄儿童实行脾脏次全切,以减轻溶血和贫血,同时保存部分脾脏的免疫功能。但是国内外均有报道,脾次全切或脾栓塞后,部分病例脾脏快速再生,复又发生严重溶血,需再次行脾全切手术。由于对脾次全切远期疗效仍缺乏足以说明问题的统计数据,所以脾次全切尚不推荐作为HS治疗首选方法。
采用腹腔镜切脾术可减轻术后不适,减少瘢痕,较快康复,缩短住院日,减少费用。术中注意不能残留脾组织,以防残脾再生影响远期疗效。如果HS患者同时伴有严重的胆囊泥沙样结石等情况需要同时切除脾脏和胆囊,仍以开腹全切为好。
6.手术失败原因 副脾、小脾、合并症和红细胞膜蛋白缺陷性质等因素可以影响脾切除术效果。据不同研究报告统计,15%~39%的HS患者存在副脾,如果切除脾脏而不切除副脾,则切脾术对溶血的改善作用可能在术后不久或术后数年明显减弱。切脾若干年后溶血复发还应当考虑小脾原因,这是切脾术中脾组织自体移植而生成的。存在副脾或小脾的提示是外周血涂片中豪-周小体(Howell-Jolly bodies)和杯形凹痕细胞消失,网织红细胞计数升高。脾组织植入的确切证据可用放射胶体肝脾扫描或用Cr标记的红细胞扫描得到证实。
HS并存其他红细胞疾病特别是有原位溶血的遗传疾病如β珠蛋白生成障碍性贫血时,由于脾切除仅去除血管外溶血主要场所,原位溶血依然存在,所以手术疗效不如单纯HS。
分析红细胞膜蛋白缺陷性质对脾切除术效果的影响,结果显示,缺乏带3蛋白的HS切脾效果不如其他膜蛋白缺乏的HS,可能的解释是:其他膜蛋白缺乏导致垂直连接异常,丢失的微囊泡含有带3蛋白,而带3蛋白与细胞变形性、细胞衰老相关,这些患者术后可减少含band 3的微囊泡丢失。而带3蛋白原发缺乏的HS患者,贫血和球形化程度相对较轻,脾脏切除后,其膜的变形功能同切脾之前相比差别不大,故临床改善效果不如其他膜蛋白缺乏的HS那样对比明显。
(二)输血
HS新生儿期溶血黄疸患儿输血1~2次后,多数不再依赖输血。有些新生儿依赖输血,可能是出生第一年对红细胞生成应激不足。原则上应尽可能避免反复输血。近年文献提倡,血红蛋白在50~60g/L或以上的较大年龄儿童无需输血,即使输血,血红蛋白维持在90g/L即可,以利于激发患者红细胞生成,避免骨髓对贫血的应激反应受到抑制。
HS红细胞膜缺失,在储存条件下会进一步脱落膜微囊泡而减少膜表面积,这种细胞的寿命明显缩短,所以HS患者在手术过程中输注自体红细胞是无益的。
(三)药物辅助治疗
目前尚无有效的HS对因治疗药物,对症治疗原则在溶血危象期为输血、输液、抗感染、防治休克与急性肾衰竭;在慢性期为补充造血原料、服用膜稳定剂,预防巨幼细胞贫血危象。
(1)抗生素类:防治感染,根据患者感染类型和药敏试验选择抗生素,重点预防荚膜菌感染尤其是肺炎球菌性败血症。脾切除术前10岁以下儿童应予接种肺炎双球菌三联疫苗,术后抗生素预防感染。①长效青霉素(苄星青霉素):成人60~120万单位,30kg以下儿童30~60万单位肌注,1次/月。②口服青霉素:7岁以下儿童125mg/次,2次/日;7岁以上儿童及成人250mg/次,2次/日。
(2)抗血栓药物:脾切除术后血小板≥800×109/L时应用,老年患者和原有心血管疾病患者需监控出凝血指标,调整用药。①阿司匹林(乙酰水杨酸)肠溶片:治疗量100mg,口服,3次/日;维持量25mg,口服,1次/日。②潘生丁(双嘧达莫):25~50mg,口服,3次/日。
(3)造血原料类维生素:①叶酸(辛帕斯):治疗量5~10mg,口服,3次/日;维持量5mg,口服,1次/日。②维生素C:口服100mg,3次/日。③维生素B12(甲钴胺,弥可保):肌注0.5mg,1次/周(必要时);口服250μg~500μg,2次/日。
(4)膜稳定剂:①维生素E(生育酚):婴幼儿10mg,1~2次/日;成人100mg,1次/日。②阿魏酸钠(川芎素片):50mg,口服,3次/日。
(5)胆红素转化剂:①苯巴比妥(鲁米那片):60mg,口服,1次/晚;(急性溶血期)成人100mg(儿童5mg/kg),必要时肌注或用高渗葡萄糖溶液配成1%浓度,缓慢静注,必要时4~6h可重复注射。②人血白蛋白:成人10~20g,儿童5~10g,必要时缓慢静滴(静滴速度不超过2ml/分)。
(6)碱化剂:碳酸氢钠、乳酸钠,碱化血液、尿液,急性溶血期防治酸中毒。
(7)能量补充剂:等渗葡萄糖液等,为重症病人补充热量,并促进有毒物质、化学药物、病菌毒素排泄,同时维持正常尿量,维护肾功能,利于纠正酸中毒。
(8)神经营养药物:伴有下肢溃疡、皮炎者,可加用维生素B6、维生素B12。
(9)激素:早期、大量、短程应用糖皮质激素可减轻急性溶血期并发症。
(10)红细胞生成素(EPO):最近国外文献推荐,9月龄以下的输血依赖性HS婴幼儿使用EPO可以减少甚至避免输血。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。











