



(四)受体学说以及药物与受体相互作用
1.受体与配基 大多数药物与生物细胞的某些特殊功能性大分子相互作用,改变了细胞相应成分的功能,进而触发了特定的一系列生理、生化效应。这些生物大分子就是药物作用的受体(receptor)。
受体这一概念是早在1906年由Langley提出的。他在观察烟碱和箭毒对骨骼肌的作用后,认为这些药物既未影响神经冲动,也不是直接作用于骨骼肌细胞,而是作用于神经与效应器间的中间物质,称之为接受物质(receptive substance),并认为烟碱与接受物质结合产生肌肉收缩,箭毒则与之竞争,引起肌肉松弛。1914年Dale通过实验把乙酰胆碱(Ach)受体分为毒蕈碱样(M)和烟碱样(N)受体。1913年Clark提出了受体-配体相互作用符合质量作用定律的占领学说和数学模型,为受体的研究奠定了重要基础。受体又被分为亚型,例如胆碱受体可分为M(毒蕈碱样)、N(烟碱样)受体,后者又分为N1、N2两个亚型。M受体近年来又被分为M1、M2、M3、M4、M5亚型。20世纪70年代以后,随着科学的发展,对受体、受体亚型逐渐获得了深刻认识。N受体于1982年克隆成功,1992—1995年相继克隆了阿片受体的δ、μ、k亚型。受体可位于细胞膜,例如单胺神经递质的受体等;也可位于细胞质内,例如甾体激素的受体等。受体是生物进化过程中形成并遗传下来的,大多数受体是蛋白质,在体内有特定的分布点。此外,受体应具有下列特征:有内源性配体、与配体结合具有高度的选择性、高亲和性、可逆性以及饱和性。配体(ligand)系指能与受体特异结合的具有生物活性的物质。机体内有内源性配基,如神经递质、激素及自体活性物质等。与受体特异性结合的外源性化学物质称为外源性配基(包括药物)。受体的数量可受疾病或与配体相互作用而变化。例如哮喘病人长期应用β-受体激动药,β-受体的数量可减少(向下调节,down regulation);反之长期应用β-受体阻断药,则β-受体的数量可增加(向上调节,up regulation)。受体向上调节或向下调节可能是药物超敏或脱敏的原因之一。
受体不但位于突触后膜,而且也可以位于突触前膜。突触前膜的受体具有调节神经递质释放的作用。例如肾上腺素能神经末梢与效应器细胞构成的突触间隙中的去甲肾上腺素浓度减少时,去甲肾上腺素可激动突触前β2受体,递质释放增加;反之,当突触间隙中的去甲肾上腺素浓度增加时,去甲肾上腺素激动突触前的α2受体,递质释放减少。去甲肾上腺素和肾上腺素还可激动豚鼠肠神经丛突触前膜的α2受体,使乙酰胆碱释放减少。
孤儿受体(orphan receptor)系指尚没有确定其配体和功能的受体。
2.受体类型、信息传递 受体不但能识别特异的配体并与之特异性结合,还能在靶细胞内转导信息,或直接引起靶细胞的效应或促进合成释放第二信使而产生效应。
(1)根据受体的生理功能、作用分类
1)离子通道受体:受体的多种亚单位跨越细胞膜组成离子通道,激动药与其结合影响离子通道的开放与闭合。例如烟碱样受体、γ-氨基丁酸(GABAA)受体、谷氨酸受体、甘氨酸受体以及5-羟色胺受体等。
2)G蛋白耦合受体:G蛋白是一类具有特异的GTP结合位点,并能水解GTP,其活性受GTP调控的蛋白。此类大多数受体位于细胞膜。激动药与其受体(如毒蕈碱样、肾上腺素、多巴胺、5-羟色胺以及阿片受体等)结合引起受体活化,与膜内侧特种G蛋白结合而引起特定效应。
3)具有酪氨酸激酶的受体:此类受体调节机体细胞的生长、分化及发育。属此类的有胰岛素受体、内皮生长因子受体以及血小板生长因子受体等。
4)激素细胞内受体:该受体位于细胞内,是可溶性DNA结合蛋白,调节特殊基因的转录,例如甾体激素受体等。
(2)信息的传递:经受体转导的跨膜信息传递的环节包括识别、转导和引起效应。即受体可以识别具有一定立体构型的配体并与之结合,经过一系列的信息的转导导致相应细胞效应器的活性变化,最终细胞产生生理活动。
受体跨膜信息转导目前大致分为:
1)配体与受体结合后改变离子通道的活性:离子通道是受体的组成成分,例如烟碱样受体、GABAA受体以及甘氨酸受体等。当激动剂与受体结合后离子通道开放、细胞膜通透性增加。另外,还有与受体耦联的离子通道,这些离子通道虽不是受体的组成成分,但通道的活性受受体的调控。受体被配体激活后,激活G蛋白,后者又可调节腺苷酸环化酶、cGTP磷酸二酯酶、磷脂酶C等,释放cAMP、二乙酰甘油(DAG)或1,4,5-三磷酸肌醇(IP3)等,从而影响离子通道的开放,cAMP、cGMP、DAG、IP3等皆为第二信使。
阳离子通道(如烟碱样受体―Na+通道)入口处的氨基酸多带负电荷;反之;阴离子通道(GABAA受体―C1¯通道)则多带正电荷。
2)通过G蛋白的调节效应的受体:属此类的受体最多,遍布机体的各个组织、器官,其激动剂包括生物胺、蛋白激素、多肽激素、花生四烯酸、淋巴活化因子、光、嗅觉等等。
G蛋白种类繁多,但它们都是膜蛋白,都由3个不同的亚单位组成:α、β、γ亚单位。α亚单位具有特异的GTP结合位点,有GTP酶活性,β、γ亚单位组成二聚体。不同的G蛋白其结构差异主要表现在α亚单位,例如兴奋性G蛋白(Gs)可激活腺苷酸环化酶(AC),只能被霍乱毒素催化,抑制性G蛋白(Ci)抑制AC,只能被百日咳毒素所催化。
当无激动剂存在时,G蛋白3个亚单位呈聚合状态(αβγ),α亚单位与GDP结合形成αβγ□GDP。当有激动剂存在时,受体(R)与其激动剂(H)相结合(RH),受体被活化并与上述三聚体形成复合物同时释放出GDP,即形成RH□αβγ。在Mg2+存在下,GTP与α亚基上原GDP位点相结合,并使复合物解离成三个部分,即R、βγ二聚体以及被激活的α□GTP亚单位,后者可激活效应器例如AC激活使cAMP合成增加,cAMP激活cAMP依赖的蛋白激酶,后者再激活它的特异底物(脂肪酶、糖原合成酶等)发挥效应。不同G蛋白可分别影响腺苷酸环化酶、cGMP磷酸二酯酶、磷脂酶C或者离子通道等。
3)具有酪氨酸激酶活性的受体:包括多肽激素(如胰岛素受体)和生长因子受体。当激动剂与细胞膜外侧受体相结合,细胞膜内的酪氨酸激酶被激活,受体自身磷酸化,然后使效应器蛋白的酪氨酸残基被磷酸化,从而改变效应器的活性。
4)激素细胞内受体:甾体激素、维生素D和甲状腺等,它们的受体位于胞质内。受体的激动剂跨过靶细胞的细胞膜与其胞质受体相结合,结合后受体活化,并易位进入细胞核。
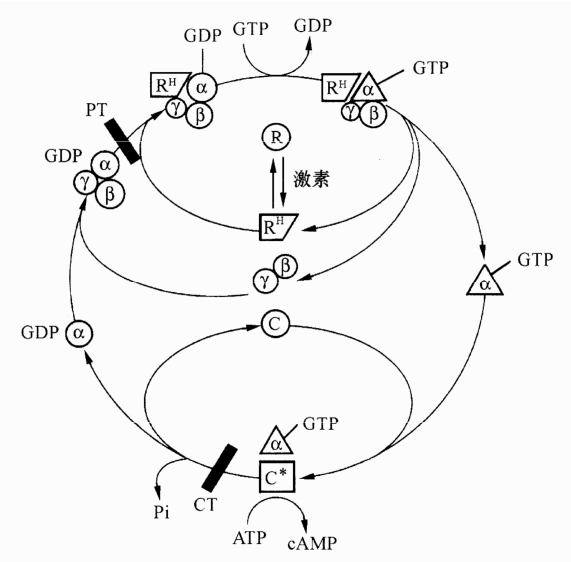
图3-5 G蛋白循环:鸟嘌呤核苷酸调节蛋白(G蛋白)循环R:无配体受体;RH:配体受体复合物;C:失活的腺苷酸环化酶催化亚基;C*:活化的腺苷酸环化酶催化亚基;α?β?γ:G蛋白亚单位;CT:霍乱毒素;PT:百日咳毒素
随后活化受体与靶基因的特定DNA序列结合,产生转录活性影响蛋白质合成而发挥作用。
3.药物与受体相互作用
(1)占领学说:20世纪初Clark提出占领学说。他认为药物的效应与药物占据的受体数量成正比。当受体被药物占据50%时,即产生50%的最大效应,此时KD=[A],KD值应等于引起50%最大效应时的药物剂量,又称半数作用量ED50,或半效浓度(ED50)。人们常用解离常数的倒数(1/KD)表示亲和力。
占领学说对受体研究有重要贡献,但它不能解释为何同系激动剂的最大效应不同,不能解释激动剂、拮抗剂的区别。20世纪50年代Ariens认为,一种药物与受体结合,不仅需要有亲和力,而且还需要内在活性。激动药(agonist)与受体有较强的亲和力,也有较强的内在活性。拮抗药(antagonist)与受体亲和力强,但缺乏内在活性。部分激动药(partial agonist)对受体亲和力不弱,但内在活性不强。Stephenson认为,对同一受体系统一个激动药引起的最大效应小于另一个高效激动药时,则前一个激动药称为部分激动药。部分激动药占据了所有能占据的受体,因内在活性低,它所引起的最大效应并非最大。当它与高效激动药同时存在时,二者竞争受体,降低了高效激动药的反应,这时部分激动药表现出拮抗药的作用。激动剂不需要占据全部受体即可引起最大效应,多余的受体即为储备受体(spare receptor)。
20世纪80年代中期发现反向激动剂(inverse agonist)存在,如苯二氮 受体拮抗剂carboline等,单用能产生与激动剂完全相反的药物作用,如动物表现为焦虑和惊厥。
受体拮抗剂carboline等,单用能产生与激动剂完全相反的药物作用,如动物表现为焦虑和惊厥。
(2)两态模型学说:受体在和配体结合之前存在两种构象即活化态和静息态,两态处在动态平衡,配体可诱导受体发生构象改变,激动剂与活化态受体结合产生效应,促进静息态转化为活化态,拮抗剂与静息态受体相结合不产生效应,但可促使活化态受体转化为静息态。两态模型学说多用来描述离子通道耦联受体。
竞争性拮抗药(competitive antagonist)与激动药作用于同一受体,两者竞争性地与受体结合,结合是可逆的。在竞争性拮抗药的存在下,激动药的最大效应不变,激动剂的量-效曲线平行右移,效价强度减小,亲和力减小。
pA2是衡量竞争性拮抗药的效价强度常用的参数,其含义是使某激动药(A)的剂量提高两倍,效应仍达到原水平所需拮抗药克分子浓度的负对数。
pA2值等于拮抗药克分子浓度的负对数,因此pA2值越大,所需的拮抗药克分子浓度越小,拮抗药的效价强度越大。又pA2值等于拮抗药平衡解离常数(K1)的倒数(亲和力)的对数,因此pA2越大,拮抗药对受体的亲和力越大,K1越小。
非竞争性拮抗药(noncompetitive antagonist)与激动药作用于不同受体,或作用同一受体,但拮抗药与受体的结合呈难逆性质。在拮抗药存在下,激动药的最大效应降低,但亲和力不变。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。









