



二、配体受体分子对接技术
(一)配体受体分子相互作用的理论计算
第一步都是将配体对接到受体的结合部位,形成配体受体复合物,这一过程称为分子对接(docking)。分子对接的目的,一是为精确的理论计算(如FEP、QM/MM等计算)提供配体受体复合物的初始构型;二是可以根据匹配情况,对配体进行结构修饰,提高配体与受体的亲和性,如配体是多肽,可以改变肽链骨架,设计非肽和类肽配体,以提高生物利用度,或将柔性配体环化,降低配体受体结合时的熵损失。
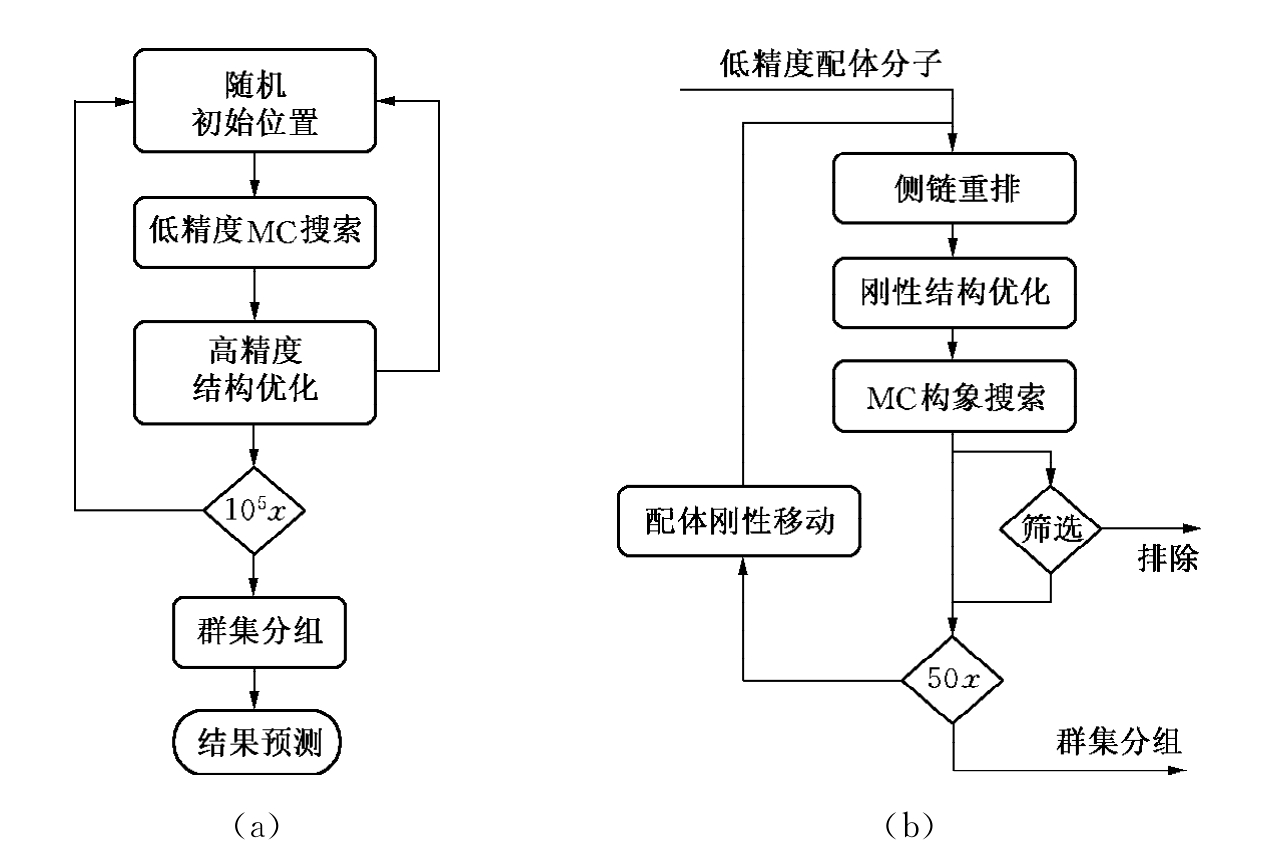
图18-6 分子对接的基本步骤
(a)对接流程图;(b)结构搜索与优化细节
(二)分子对接的一般流程
通常可以分为以下基本步骤(图18-6):
(1)低精度构象搜索。
(2)对搜索得到的构象库利用能量函数进行打分、分类和筛选。
(3)对高分构象进行结构优化并作能量评价。
(4)最后得到模拟的配体受体复合物结构。
(三)分子对接的类型
分子对接根据条件和目的的不同,分为实时图形对接(interactive molecular graphics ap-proaches)和自动对接(automatic docking)2种。
1.实时图形对接 以已知配体受体复合物的晶体结构为基础,计算受体结合部位的各种表面性质(静电、氢键和疏水势等的分布),删去原有的配体,计算分子对接配体的表面性质,然后根据互补匹配规则,将配体对接到受体的结合部位。现有的图形工作站(如SGI)和分子模拟软件(如SYBYL和insightⅡ)使这一过程非常容易进行。
实时图形处理途径在以结构为基础的药物设计中有广泛的应用,并取得了很大的成功。实时图像处理的优点是能在计算机屏幕上形象直观地显示并操作分子间的相互作用过程;实时图形处理可以解释构效关系,解决3D-QSAR研究中对活性构象的需要;此外,还能与自动对接方法配合使用。
2.自动对接 在无人工干预的情况下实现配体和受体的对接。首先识别蛋白质表面的结合部位,一般为状似口袋(pocket/cave)或沟槽(groove/cleft)样区域;然后放置配体并优化空间定位。美国加州大学旧金山分校Kuntz研究组开发的DOCK程序是自动对接方法的典型代表。该程序包括3部分:①配体和受体表面结构的表达。②配体和受体表面结构的匹配。③配体取向和位置优化。
(四)分子表面的球形表达
采用Connolly提出的MS算法,能精确表达分子表面形状,Connolly表面由探针原子在分子的范德华表面上滚动而产生。Connolly表面由接触表面(contact surface)和凹面(reentrant sur-face)2部分组成(图18-7),与范德华表面相比,Connolly表面是连续、光滑的。有关MS算法和受体结合部位的寻找方法,可查阅相关的文献。
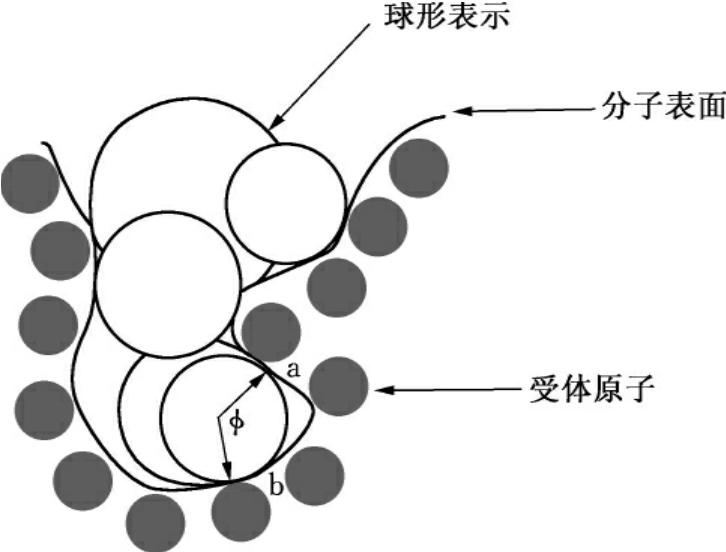
图18-7 受体分子表面球形表示
(五)集团匹配(clique matching)
得到上述受体结合部位和配体表面的球形表示后,必须利用球形表示将配体和受体对接。分子对接时,将配体和受体的球形一一配对,配体分子平动和转动6个自由度的变化通过配体球和受体球的配对来体现,在配对时,必须调整配体的方向,找到最佳位置。如尝试每一种配对的可能性,计算量非常大,因为,如配体有m个球形组成,受体结合位点有n个球形组成,则所有配对的可能数目为n!/(n-m)!如果ns=50,nl=25,则配对可能性约为1039,这是系统测试所不允许的。DOCK程序为克服这一困难,采用集团匹配方法进行分子对接。假设已有配体球I与受体球K匹配,则下一个匹配对j和l应当与尽可能多的已有匹配对(j∈I,l∈K)满足以下条件
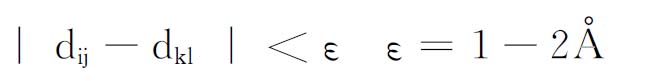
至少要找到4对匹配球形,才能决定配体的结合方位。通常,这种方法可使配对可能性降到几千以内,大大提高了分子对接的速率。如仅考虑形状匹配,通常用SGI机器每分钟可对接2个配体。
上述分子对接,仅考虑分子的形状匹配,并且在对接过程中配体和受体保持刚性以减少自由度,这在搜寻数据库时会失去很多机会。如在剑桥晶体结构库(CSD)中,二氢叶酸还原酶(DHRF)抑制剂甲氨蝶呤存在2种构象的晶体结构,但均不是与DHRF结合时的构象,所以用刚性分子对接方法搜寻数据库有其局限性。
此外,早期的自动对接方法仅用Eoverlap作为评价函数,Eoverlap表征配体和受体范德华半径的重叠误差为:
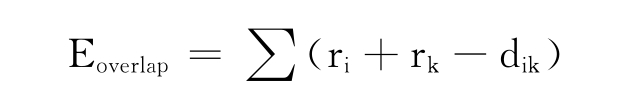
近年来对分子对接方法进行了改进,DOCK程序引入了分子力场作评价函数,在分子对接时考虑或部分考虑分子的柔性,并且药物与受体结合的打分函数也考虑了静电、氢键和疏水等相互作用。
以DOCK程序为代表的自动对接技术的一个重要用途是进行分子对接数据库搜索,结合高性能计算机和并行计算用于高通量的药物虚拟筛选。
美国南卡来罗纳医学院Ilya AVakser教授开发的GRAMM程序提供了另一种基于配体受体分子表面形状互补性和疏水作用的自动对接方法,其中的疏水作用对接方法最为简单有效,尤其适用于蛋白质分子之间的相互作用。
上述两种分子对接均可归类为已知配体对接方法,除此之外,在配体结构未知的情况下,直接在受体结合部位构建或生成与之相匹配的化合物,这种方法称为全新配体对接或全新药物设计(de novo drug design)。
全新配体对接设计方法按其所用的构建模块(building block)不同,分为3类:①模板定位法;②原子生长法;③分子碎片法。其中以分子碎片法为主,而后者又以德国BASF实验室设计的LUDI程序为代表。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。









