



2.1 材料的内部结构
绝大多数机械工程材料的使用状态为固态,固态材料的内部结构即构成材料的原子(或离子、分子)在三维空间的结合和排列状况(聚集状态),可分为三个层次,如图2-1所示:一是组成材料的单个原子结构和彼此的结合方式(金属键、离子键、共价键、分子键);二是原子的空间排列;三是宏观与微观组织。材料的性能除与其组成原子或分子的种类有关外,主要取决于它们的聚集状态,即材料的内部结构。

图2-1 材料不同层次的结构示意图
2.1.1 金属的结构
1.晶体与非晶体
所有的固态物质,就其原子(或分子)排列的规则性来分类,可分为晶体和非晶体两大类。固态物质内部的原子(或分子)呈周期性规则排列的称为“晶体”,如水晶、天然金刚石、食盐等;否则,为“非晶体”,如石蜡、松香等。大部分液态物质凝固时,它们黏度很低,原子或分子易扩散聚集成稳定的晶体状态,如金属材料;而高黏度的熔体凝固时则常形成非晶体,如多数高聚物及玻璃材料。晶体具有固定熔点,各向异性(指单晶)等特征;而非晶体无固定熔点,它是在一个温度范围内熔化,各方向上原子聚集密度大致相同,所以表现为各向同性。在非晶态结构中,原子排列没有规则的周期性,即原子的排列从总体上是无规则的(即远程无序),但是,近邻原子的排列是有一定规律的(即近程有序);例如,非晶硅的每个原子仍为四价共价键,与最邻近原子构成四面体,这是有规律性的;而总体原子的排列却没有周期性的规律,呈玻璃态物质。晶体与非晶体在一定条件下可以互相转化。
图2-2所示为一种最简单的晶体结构示意图,原子按简单立方体的方式堆垛,构成简单立方晶体结构。自然界的固态物质尤其是金属大多数属于晶体,具有特定的晶体结构。晶体的性能在很大程度上由其晶体结构(原子,离子或分子的排列方式)所决定。
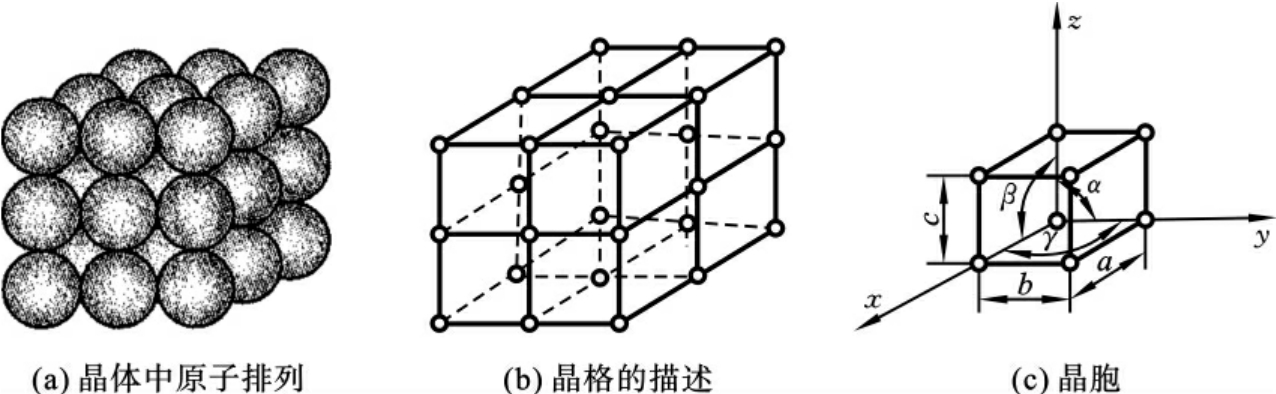
图2-2 简单的晶体结构示意图
为了便于研究和分析晶体中原子排列的规律性,通常采用经典的刚球模型,即将晶体中的原子或正离子看作刚球,假设晶体是由许多刚球按一定几何规律紧密堆垛而成的。为了分析堆垛的规律,将原子或正离子抽象为一个几何点,其位置代表原子中心所在的位置,然后用假想的直线将所有代表原子的几何点连接起来,构成三维空间的几何晶格架(见图2-2(b))。这种描述原子在晶格中排列形成的空间格子,通常称为“晶格”;构成晶格的各连线的交点称为“结点”,显然,结点是一个几何点,它表示一个原子中心的位置。
由于晶体中原子排列具有一定的规律,由图2-2可以看出,晶格形状特征反映出该晶格中原子排列形式的规律。组成晶格的、能反映晶格特征的最基本的几何单元称为“晶胞”(见图2-2(c)),而晶胞在三维空间的重复排列构成晶格并形成晶体。组成晶胞的各棱边的尺寸a、b、c称为“晶格常数”,金属的晶格常数一般在1~7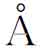 (1
(1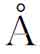 =10-10m);各相邻棱边之间的夹角分别用α、β、γ表示。通常,在晶胞上取左下方后面的结点作为坐标原点o,取晶胞中交于点o的三个棱边为坐标轴x、y、z,晶胞的形状可以由a、b、c和α、β、γ六个参数决定。参数不同,反映出晶格的类型不同,例如在图2-2中,a=b=c,α=β=γ=90°,这种晶胞称为“简单立方晶胞”,其晶格为“简单立方晶格”。在晶体学中,通过晶体中原子中心的某一方位的原子面称为晶面,而通过原子中心的某一方向的原子列称为晶向。金属的晶体结构可用X射线结构分析技术进行测定。
=10-10m);各相邻棱边之间的夹角分别用α、β、γ表示。通常,在晶胞上取左下方后面的结点作为坐标原点o,取晶胞中交于点o的三个棱边为坐标轴x、y、z,晶胞的形状可以由a、b、c和α、β、γ六个参数决定。参数不同,反映出晶格的类型不同,例如在图2-2中,a=b=c,α=β=γ=90°,这种晶胞称为“简单立方晶胞”,其晶格为“简单立方晶格”。在晶体学中,通过晶体中原子中心的某一方位的原子面称为晶面,而通过原子中心的某一方向的原子列称为晶向。金属的晶体结构可用X射线结构分析技术进行测定。
2.纯金属的晶体结构
由于金属晶体中的原子是金属键结合,使原子具有趋于紧密排列的倾向,因而常常形成几种高度对称的、几何形状简单的晶格。在元素周期表中,90%以上的金属元素的晶体都属于以下三种原子紧密排列的晶格形式。
1)体心立方晶格(body-centred cubic lattice,bcc)
如图2-3所示,体心立方晶格的晶胞是由8个处于顶角结点位置的原子构成的一个立方体,在该立方体的中心位置上还有一个原子,其晶胞参数为a=b=c,α=β=γ=90°,属于立方晶系。在该晶胞中,体对角线方向上的原子排列紧密接触,由此可以求出原子半径r与晶格常数a的关系为r=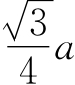 。由于每个顶角上的原子为周围相邻8个晶胞所共有,每个晶胞只占有其1/8,而体心位置的原子为该晶胞所独有,因此,一个体心立方晶胞中含有
。由于每个顶角上的原子为周围相邻8个晶胞所共有,每个晶胞只占有其1/8,而体心位置的原子为该晶胞所独有,因此,一个体心立方晶胞中含有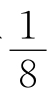 ×8+1=2,即2个原子。
×8+1=2,即2个原子。
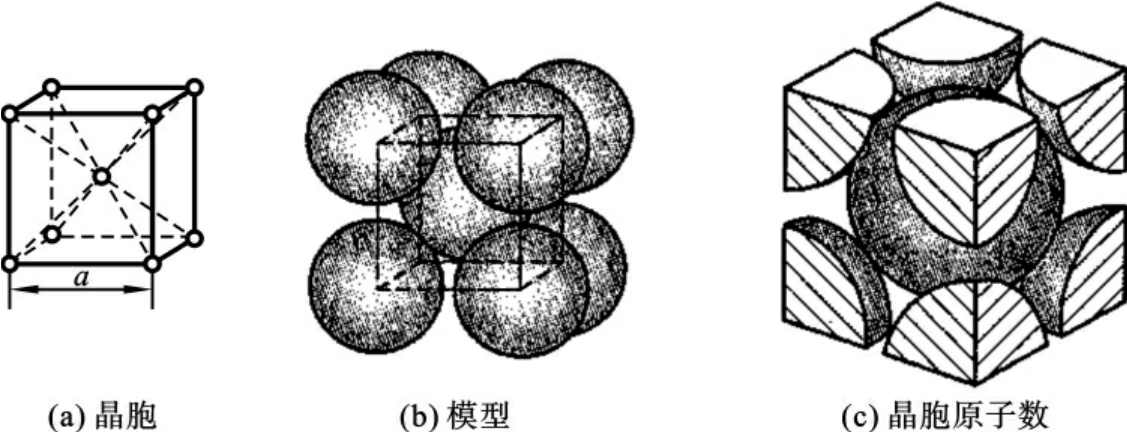
图2-3 体心立方结构晶格示意图
原子排列的紧密程度通常用晶格的“致密度”来表示。所谓致密度是指晶胞中所含有的原子实际占有的体积与该晶胞的体积之比。在体心立方晶体中,每个晶胞含有2个原子,占有的体积为2×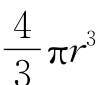 ,而晶胞的体积为a3,因此致密度为
,而晶胞的体积为a3,因此致密度为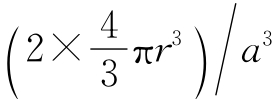 =0.68。即体心立方晶格中有68%的体积被原子所占有,其余的32%为空隙。
=0.68。即体心立方晶格中有68%的体积被原子所占有,其余的32%为空隙。
另一种表示原子排列紧密程度的方法是采用“配位数”的概念。所谓配位数是指晶格中任意一个原子周围最近邻的等距离的原子个数(或紧密接触的原子数)。配位数越大,表示原子排列得越紧密。由图2-3可见,8个顶角原子与体心原子为紧密接触,而且距离相等,所以体心立方晶格的配位数为8。
具有体心立方晶格的金属有铁(α-Fe)、铬(Cr)、钼(Mo)、钨(W)、钒(V)、铌(Nb)等,其大多具有较高熔点、硬度及强度,而塑性、韧度较低,并具有冷脆性倾向。
2)面心立方晶格(face-centred cubic lattice,fcc)
如图2-4所示,面心立方晶格也属立方晶系,在晶胞立方体的每一个构成面的中心位置还各有一个原子。该晶胞中,在每一个构成面的对角线方向上各原子彼此接触,因而原子半径r=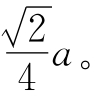 面心立方晶胞中含有4个原子,因为每个对角上的原子为8个相邻晶胞所共有,而每个面心位置的原子为2个相邻晶胞所共有,配位数为12。面心立方晶格属于原子排列最紧密的结构,其致密度为0.74。
面心立方晶胞中含有4个原子,因为每个对角上的原子为8个相邻晶胞所共有,而每个面心位置的原子为2个相邻晶胞所共有,配位数为12。面心立方晶格属于原子排列最紧密的结构,其致密度为0.74。
具有面心立方晶格的金属有铁(γ-Fe)、铝(Al)、铜(Cu)、镍(Ni)、铅(Pb)、银(Ag)、金(Au)等,大多有较高的塑性,没有冷脆性倾向。
3)密排六方晶格(hexagonal closed-packed lattice,hcp)
如图2-5所示,密排六方晶格的晶胞是由12个原子占据简单六方体的12个顶角位置,其上、下两个正六边形面的中心位置还有1个原子,而且在此六方体的中间还有3个原子。其晶胞参数a=b≠c,其中c/a(称为“轴比”)约为1.633,其每个晶胞内含有6个原子。密排六方晶格也是原子排列最紧密的结构,其致密度、配位数和面心立方晶格相同,分别等于0.74和12。
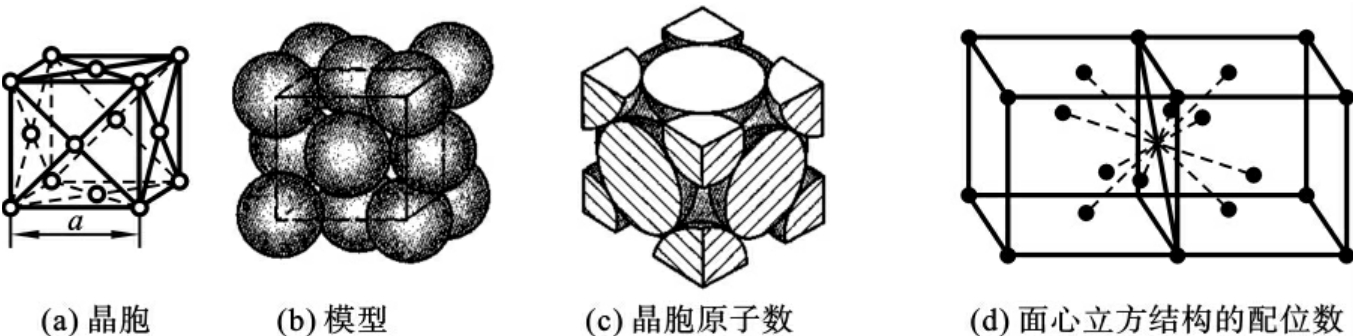
图2-4 面心立方结构晶格示意图
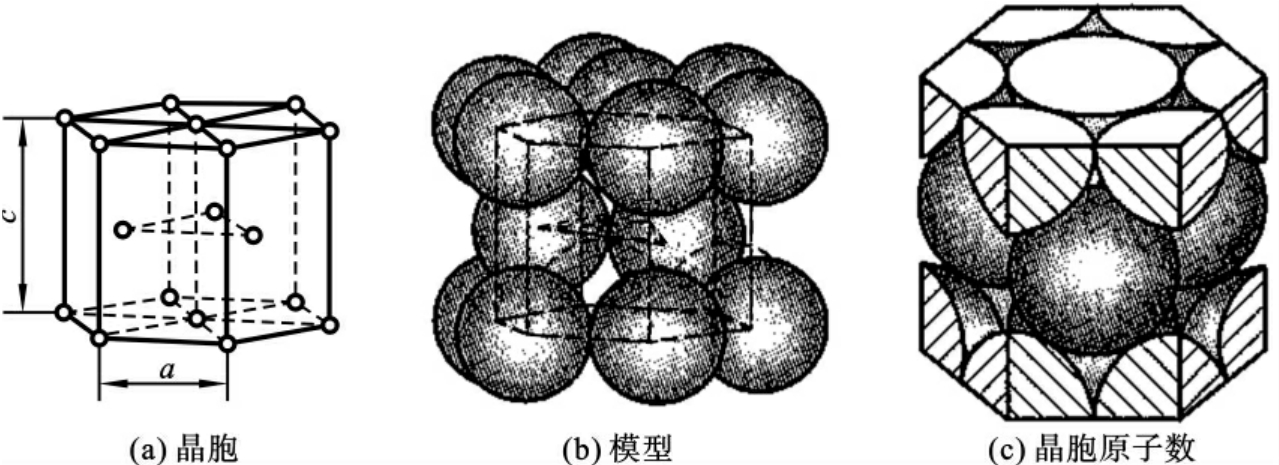
图2-5 密排六方结构晶格示意图
具有密排六方晶格的金属有铍(Be)、镁(Mg)、锌(Zn)、镉(Cd)等,石墨也是密排六方晶体结构,其大多没有冷脆性,但力学性能不突出,很少单独用于结构材料。
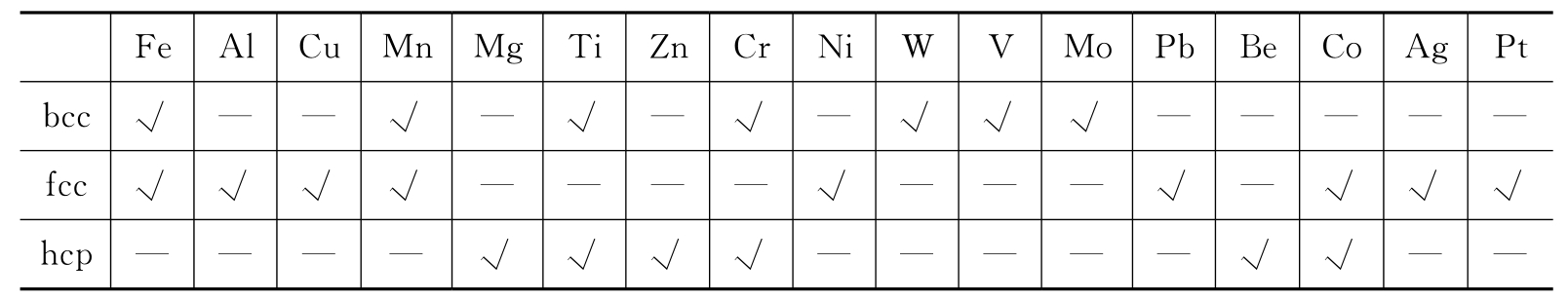
少数金属(如铁、锰、钛等)在晶态时,其晶格类型会随外界条件(如温度,压力等)而改变,常压下常用金属的晶体结构如表2-1所示。
表2-1 常压下常用金属的晶体结构
对某一元素,当其从一种晶格变为另一种晶格时,因致密度的不同,必然导致体积的变化,如α-Feγ─-Fe;另一方面,同一元素处于不同晶格类型时,其原子半径是 不同的(因已假想原子为紧密接触的刚性球体),原子排列的对称性也不同,这就导致面心立方的γ-Fe比体心立方的α-Fe空隙半径大,溶入碳的能力大。人们可利用这种现象,改变晶体结构及其特性(如钢的热处理),以实现改变材料性能的目的。
不同的(因已假想原子为紧密接触的刚性球体),原子排列的对称性也不同,这就导致面心立方的γ-Fe比体心立方的α-Fe空隙半径大,溶入碳的能力大。人们可利用这种现象,改变晶体结构及其特性(如钢的热处理),以实现改变材料性能的目的。
此外,从上述晶格的几何特征很易看出,不同晶面及晶向上原子排列的方式和密度不同,则原子间的结合力大小也不同,必然导致相应的性能差异,此即理想晶体的各向异性现象。金属的许多性能及金属中发生的许多现象都与金属晶体中的晶面和晶向有密切关系。
3.实际金属的结构
实际金属晶体内部的原子排列并不像理想晶体那样规则和完整,总是存在着一些原子偏离理想规则排列的区域,此即晶体缺陷。这些缺陷造成了实际晶体的不完整性,并对金属和含有晶体相的材料的许多性能产生极其重要的影响。晶体缺陷包括以下几个方面。
1)空位、间隙原子和置换原子
晶体中的原子在其平衡位置上作热振动,振动能量与晶体温度有关,温度愈高,能量愈大。但是,各个原子的能量并不完全相等,而是呈统计分布,并经常地变化着,于是,一些高能量的原子就有可能脱离原来的平衡位置,迁移到晶体表面或原子之间的间隙位置,使原来的位置空着,称之为空位,空位的存在利于内部原子扩散;处于间隙中的原子则称为间隙原子;占据在原来基体原子平衡位置上的异类原子称为置换原子。空位、间隙原子和置换原子(见图2-6)都破坏了晶格的规则性,造成晶体缺陷,它们呈点状不规则排列,三维尺寸很小,故又统称为晶体的点缺陷,其会导致金属的强度、电阻等增加,塑性下降,是固溶强化的主要原因。
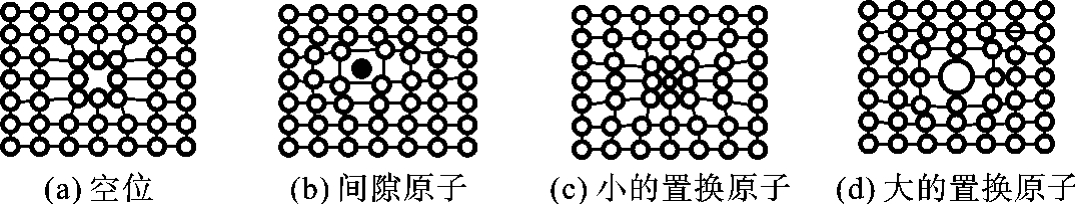
图2-6 晶体中的点缺陷
2)位错
位错是晶体中原子呈线状排列不规则的现象,又称线缺陷,由晶体中原子面的错动引起。最常见的形式是刃型位错和螺型位错,如图2-7所示。

图2-7 位错模型示意图
刃型位错是指晶体的一部分相对于另一部分出现一个多余的半原子面。这个多余的半原子面如同切入晶体的刀片,刀片的刃口线附近(此微小区域的原子排列不规则)便是位错线所在位置。
而螺型位错如图2-7(b)所示,是晶体右边上部的点相对于下部的点向后错动一个原子间距,若将错动区的原子用线连接起来,则具有螺旋形特征。
晶体中位错的数量通常用位错密度来表示,它是指单位体积晶体中所包含的位错线总长度,一般用ρ表示,单位是cm/cm3或cm-2。实际金属晶体中的位错密度与其状态有关,如不含位错(即位错密度=0)的理想单晶体金属,则强度会很高;在退火状态下,金属的位错密度为106~108cm-2,此时强、硬度低,而塑性高;而在经受冷变形的金属中,位错密度增至1011~1012cm-2,此时强、硬度将大大升高,而塑性明显下降,高密度的线缺陷是导致加工硬化的主要原因之一。
晶体位错密度可用X射线或透射电子显微镜测定。
位错线上多余原子面处于左右晶格结点临界位置,具有易动性,如图2-8所示,在不大的切应力作用下,使位错易向左或右移动到另一稳定位置,直到从晶体中移出为止,导致晶体上下相互产生一个原子间距的相对滑动(称滑移),无数的位错滑移则导致晶体产生了宏观塑性变形,这就是金属固态塑性变形的主要实质。
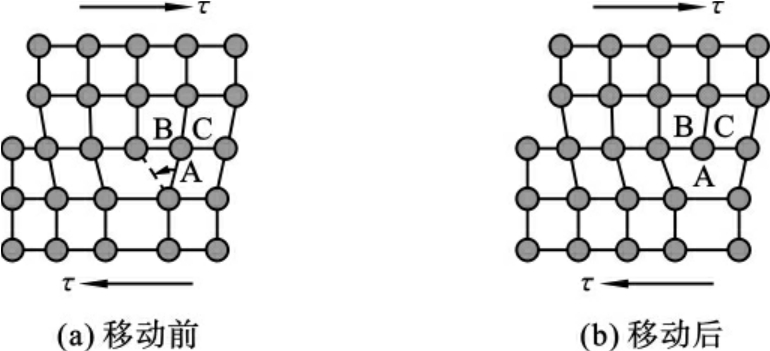
图2-8 刃型位错在切应力作用下的移动
3)晶界
如一块晶体内部的晶格位向完全一致,该晶体就称为单晶体。实际使用的金属多是由无数晶格位向不同的单晶体组成的多晶体,此时的单个晶体又称为晶粒,如图2-9所示。金属与陶瓷通常都是多晶体,由于各晶粒的位向不同,使其原子排列的规律性在相互交界处得不到统一,必须从一种排列取向过渡到另一种排列取向。晶界就是不同取向晶粒之间的过渡层,如图2-10所示,其宽度约为几个原子,其原子排列得比较不规则,晶界处还存在着许多缺陷,如杂质原子、空位及位错等。此外,在一个晶粒内部也存在一些位向稍有差别的小晶块,称为亚结构或亚晶,它们之间的界面称为亚晶界。晶界与亚晶界是具有缺陷的界面,故称之为面缺陷。
晶界处因能量较高,稳定性较差,使晶界处熔点较低,易受腐蚀等。但常温下晶界却对位错的移动有阻碍作用,而晶体的塑性变形主要是靠无数位错的滑移来进行的,显然,同样的金属材料在相同的变形条件下,晶粒越细(即相当于晶粒细化),晶界数量就越多,晶界对塑性变形的抗力越大,同时晶粒的变形也越均匀,致使强度、硬度越高,塑性、韧度也好。因此,在常温下使用的金属材料,通常晶粒越细越好。但晶界在高温下的稳定性差,则晶粒越细,其高温性能就越差。面缺陷的增加,是细晶强化的主要原因,而位错密度增加、亚结构细化则是加工硬化的主要原因。
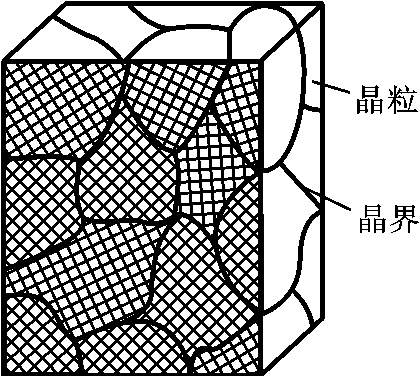
图2-9 多晶体结构示意图
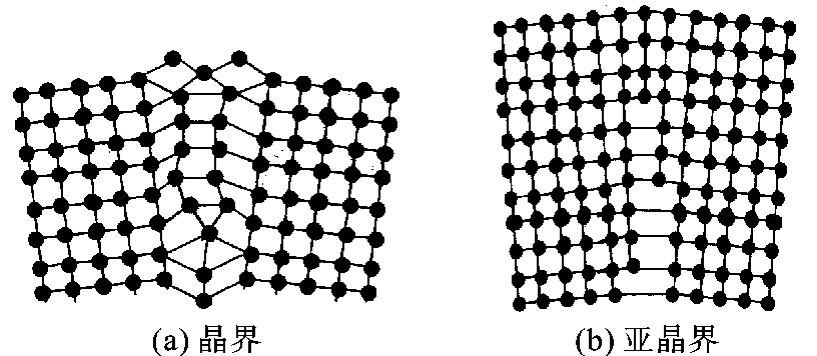
图2-10 面缺陷示意图
晶体中的点缺陷、线缺陷、面缺陷都使晶格畸变,使晶体缺陷处于高能量的不稳定状态。
4.合金的相结构
纯金属具有较好的导电、导热等理化性能,但其力学性能一般较低,且价格较高,冶炼也较难,故在工业上很少作为结构类材料使用。实际中大量使用的都是由两种或两种以上元素组成的具有金属特性的材料——合金。组成合金的独立的、最基本的单元称为组元,组元可以是元素或稳定化合物,如铁-碳合金中纯铁和碳都是组元,在黄铜中铜与锌也都是合金的组元,陶瓷材料中的组元多为化合物,如SiO2、Al2O3等。不同组元或相同组元但质量百分数不同可构成一系列的合金,称为合金系,如铁-碳合金(系)、铝硅合金(系)等。
合金中具有同一化学成分、同一晶体结构或同一原子聚集状态,并有界面分隔的各个均匀组成部分称为相。因此,凡是化学成分相同、晶体结构与性质相同的物质,不管其形状是否相同,不论其分布是否一样,统称为一个相。例如铁-碳合金结晶时,如固态与液态同时存在,则是两个相或三个相(如共晶结晶)。
由于形成条件的不同,合金中可形成不同数量、形态、大小和分布的各种相。固态合金可由单一相或多个相结构组成,即常说的单相组织或多相组织(组织是指用肉眼或显微镜观察到的合金中不同组成相的数量、形态、大小和分布的组合状态)。
由结构特点的不同,合金中的相分为固溶体和金属化合物两大类。
1)固溶体
固溶体是指溶质原子溶入固态溶剂中形成的相,固溶体保持溶剂组元所固有的晶体结构。根据溶质组元原子在溶剂结构中的分布形式,可把固溶体分为置换固溶体和间隙固溶体两种类型。
(1)置换固溶体 如图2-11(a)所示,溶质原子置换了溶剂晶格中一些溶剂原子就形成置换固溶体。当两组元在固态呈无限溶解时,所形成的固溶体称为连续固溶体或无限固溶体(此时量多者为溶剂);当两组元在固态呈有限溶解时,只能形成有限固溶体。
(2)间隙固溶体 某些原子半径很小的非金属元素,如氢(0.46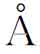 )、硼(0.97
)、硼(0.97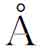 )、碳(0.77
)、碳(0.77 )、氮(0.71
)、氮(0.71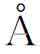 )和氧(0.61
)和氧(0.61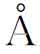 )等,溶入过渡族金属晶格间隙内,便形成间隙固溶体(见图2-11(b))。此外,当以化合物为溶剂时,也能形成间隙固溶体,例如,Ni溶入NiSb中便属于这种情况。
)等,溶入过渡族金属晶格间隙内,便形成间隙固溶体(见图2-11(b))。此外,当以化合物为溶剂时,也能形成间隙固溶体,例如,Ni溶入NiSb中便属于这种情况。

图2-11 固溶体的两种类型示意图
当两组元的原子半径和电化学特性接近,以及晶格类型相同时,易形成置换固溶体,并有可能形成无限固溶体;当组元间原子半径相差较大时,易形成间隙固溶体。间隙固溶体都是有限固溶体,其溶质的分布一般是无序的(无序固溶体)。
在有限固溶体中,溶质元素在固溶体中的极限浓度称为固溶度(即饱和浓度)。通常在高温下达到饱和的固溶体,随着温度的降低,溶质原子将从固溶体中析出而形成新相。
虽然固溶体的晶体结构和溶剂的相同,但因溶质原子的溶入引起晶格常数改变,形成点缺陷并导致晶格畸变,使位错移动阻力增加,合金的强度、硬度、电阻增高,塑性、耐蚀性降低。这种通过加入溶质元素形成固溶体,使合金强度和硬度升高的现象称为固溶强化。适当控制溶质元素的量,可以在显著提高合金强度的同时,又保持较高的塑性和韧度。因此,对综合力学性能要求高的零件材料,大都采用以固溶体为基体的合金。
在三元或三元以上合金中,可能同时兼有上述两种形式的固溶体。
2)金属化合物
金属化合物是指金属与金属元素之间或金属与类金属(以及部分非金属)元素之间的化合物。这些化合物的晶体结构与其组元的晶体结构完全不同。一部分金属化合物的成分还可在某个范围内变化,从而使其兼有固溶体的特征。金属化合物中除有离子键或共价键外,还有部分金属键,使其具有一定程度的金属特性,如导电性、金属光泽等,因此称为金属化合物。
金属化合物的类型很多,一般分为正常价化合物(如Mg2Si、MnS、Mg2Sn等)、电子化合物(如CuZn、Cu3Al等)和间隙化合物(如Fe4N、VC、WC、TiN、Fe3C、Cr23C6等)三大类。它们的晶体结构除有前述的三种晶格外,还有一些是各种复杂晶体结构,如铁-碳合金中的渗碳体(Fe3C)是一种由铁元素和碳元素组成的具有复杂结构的间隙化合物。
金属化合物一般具有较高的熔点、高的硬度和较大的脆性。合金中出现金属间化合物时,可提高材料的强度、硬度和耐磨性,但是塑性降低。适当数量与分布的金属化合物可作为强化相,如在固溶体基体上弥散分布适当的金属化合物,是导致合金材料产生弥散强化(或沉淀强化)的原因。
由两种及以上固溶体或金属化合物混合在一起而形成的多相固体组织称为机械混合物,如铁-碳合金中的珠光体(P)、莱氏体(Ld)等,其性能取决于各组成相的种类、数量、形态、大小、和分布状况。
2.1.2 有机高分子材料的结构
虽然有机高分子物质的相对分子质量大,并且结构复杂多变,但组成高分子的大分子链都是由一种或几种简单的低分子有机化合物以共价键重复连接而成的,就像一根链条是由众多链环连接而成一样。高分子化合物的结构大致可分为单个大分子链结构和大分子间的聚集态结构。
1.单个大分子链的结构
凡是可以聚合生成大分子链的低分子化合物称为单体,如聚氯乙烯可看成是由数量足够多的低分子氯乙烯聚合而成的,氯乙烯(CH2═CHCl)就是聚氯乙烯的单体,写成反应式为
n(CH2═CHCl)─→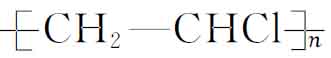
其中大分子链中的重复结构单元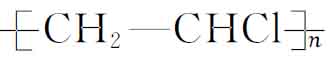 称为链节,—Cl为取代基,n为一个大分子链中链节的重复次数,即称为聚合度。聚合度越高,分子链越长,分子链的链节数就越多。聚合度反映了大分子链的长短和相对分子质量的大小,但由于各个分子的聚合度的不同,所以一般所指的高聚物分子量为平均相对分子质量。相对分子质量的分散性对高聚物性能会产生一定的影响。
称为链节,—Cl为取代基,n为一个大分子链中链节的重复次数,即称为聚合度。聚合度越高,分子链越长,分子链的链节数就越多。聚合度反映了大分子链的长短和相对分子质量的大小,但由于各个分子的聚合度的不同,所以一般所指的高聚物分子量为平均相对分子质量。相对分子质量的分散性对高聚物性能会产生一定的影响。
大分子链中的原子或原子团在空间的排列形式称为空间构型,有如图2-12所示的三种。如取代基在大分子主链上前后排列顺序不同,或者在大分子主链两侧排列的位置不同,均会对高聚物性能产生影响。如取代基—CH3在主链两侧作不规则分布的所谓无规立构聚丙烯在室温时为液体,而取代基—CH3全部在主链一侧的所谓全同立构聚丙烯则可作塑料和纤维。此外,主链侧取代基的大小不同、极性不同,均会对性能产生很大影响。
由低分子化合物合成为高分子化合物的反应称为聚合反应,其有加聚和缩聚两种类型。
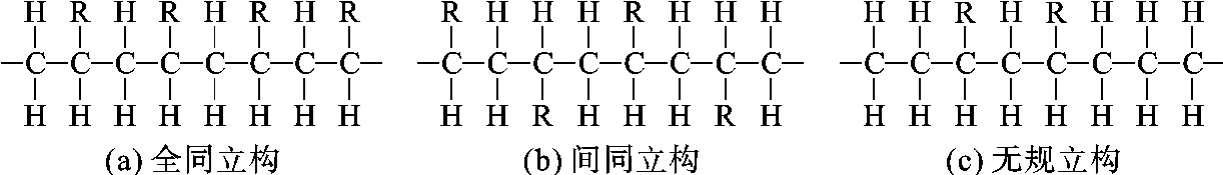
图2-12 乙烯类聚合物的空间构型
2. 大分子链的形状
单个高分子化合物的形状,如图2-13所示。

图2-13 高分子链的形态示意图
(1)线型分子结构是由许多链节组成的长链,通常是卷曲线形。具有这类结构的高聚物弹性、塑性好,硬度低,常可被溶剂溶解和受热熔化,为“可溶可熔”,如热塑性塑料。
(2)支链型分子结构在主链上带有支链。由于支链的存在,使分子链不易形成规则排列,分子之间作用力下降,分子链易卷曲,从而提高了高聚物的弹性和塑性,降低了结晶度、成形加工温度及强度。
(3)体(网)型分子结构的分子链之间有许多链节以共价键互相交联。这类结构的高聚物硬度高,有一定耐热性及化学稳定性,脆性大,无弹性(橡胶除外)和塑性,常为“不溶不熔”,如热固性塑料。
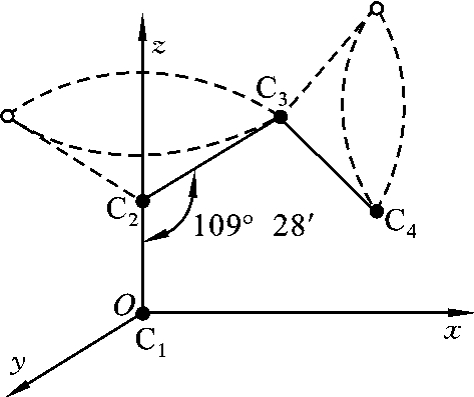
图2-14 C—C键的内旋转示意图
3.大分子链中单键内旋转和链的柔顺性
大分子链的主链都是通过共价键连接起来的,它有一定的键长和键角,如C—C键的键长是0.154 nm,键角为109°28′,在保持键长和键角不变的情况下它们可以任意旋转,这就是单键的内旋转,如图2-14所示。
单键内旋转的结果,使原子排列位置不断变化。大分子链很长,由于热运动,每个单键都在内旋转,而且频率很高(如室温下乙烷分子可达1011~1012Hz),就必然造成大分子的微观形态瞬息万变。这种由于单键内旋转所引起的原子在空间占据不同位置所构成的分子链的各种形象,称为大分子链的构象。大分子链的空间形象变化频繁,构象多,就像一团随便卷在一起的细钢丝一样,对外力有很大的适应性,即受力时可以表现出很大的伸缩能力。大分子这种因单键内旋而改变其构象,从而获得不同卷曲程度的特性称为大分子链的柔顺性,这也是聚合物有弹性的原因。
当大分子主链全部由单键组成时,分子的柔顺性差;当主链中含有芳杂环时,柔顺性也差;主链侧的侧基极性大、体积大时,柔顺性也差;温度升高时,分子热运动加剧,柔顺增加。
4.高分子的聚集态
线型高分子在分子间力作用下的聚集状态有晶态(分子链在空间规则排列,如折叠状或平行状等),部分晶态(分子链在空间部分规则排列)和非晶态(分子链在空间无规则排列,亦称玻璃态或无定形态)三种,如图2-15所示。通常线型聚合物在一定条件下可以形成晶态或部分晶态,而体型聚合物只能为非晶态或称玻璃态(因仅为一个体网型大分子,无聚集状态可言)。在实际生产中获得完全晶态的聚合物是很困难的,大多数聚合物都是部分晶态或完全非晶态。通常,用聚合物中晶态区域所占的重量或体积百分数即结晶度来表示聚合物的结晶程度,聚合物的结晶度变化范围很宽,为30%~90%,特殊情况下可达98%,而一个大分子链可以同时穿过多个晶区和非晶区。

图2-15 高聚物聚集状态示意图
一般情况下,结晶度高的高聚物,其强度、硬度、密度、耐热性、耐蚀性均较高,但弹性、塑性、透明性则有所下降。主链的结构、侧基的体积和极性、是否受定向拉伸,以及冷却速度的快慢均对结晶度有影响。
因高聚物相对分子质量很大,所以各分子链之间的分子间力的合力常远大于主链共价键力,使高聚物极易凝成固体或高温熔体,而不存在气态,若温度过高,则直接分解。
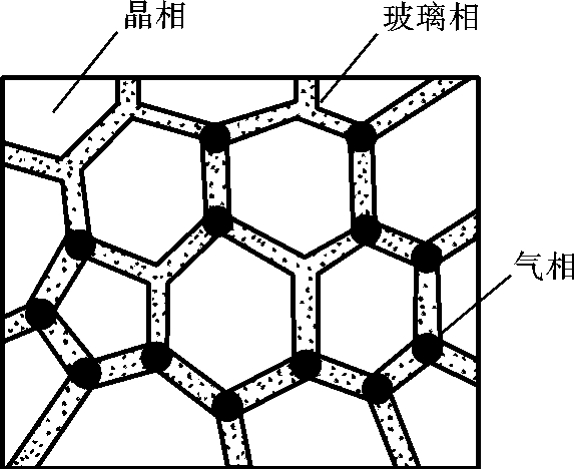
图2-16 陶瓷的组织结构示意图
2.1.3 陶瓷材料的结构
一般陶瓷材料均为多元系,其组成相可分为固溶体和化合物两大类,但其具体内容和组织组成物要比金属的金相组织复杂得多。
1.陶瓷材料的相组成
陶瓷材料中除了晶体相外,还有非晶体的玻璃相和气相,如图2-16所示,它们对陶瓷材料的性能均起重要的作用。
1)晶体相
与金属材料相似,陶瓷材料通常由多种晶体组成,有主晶相(量多的晶相)、次晶相及第三晶相等。部分晶相在不同温度下还会发生同素异晶转变。不同成分、不同工艺得到的陶瓷组织各不相同。主晶相的性能往往标志着陶瓷的物理化学性能。陶瓷中晶体相常为硅酸盐、氧化物和非氧化物三种。
2)玻璃相
陶瓷材料内各种组分和混入的杂质在高温烧成时发生物理、化学反应,常会形成低熔点的或高黏度的液相,冷却后便可能以玻璃相(非晶态)形式出现。玻璃相的主要作用是:在瓷坯中起黏结作用,把分散的晶相黏结在一起;起填充气孔空隙作用,使瓷坯致密化;降低烧结温度;抑制晶粒长大等。玻璃相所占比例一般为20%~40%;玻璃相过多,陶瓷的熔点将降低。
3)气相
气相是指陶瓷组织内部残留下来的孔洞。通常残留气孔量为5%~10%,特种陶瓷在5%以下。陶瓷材料的性能与气孔的含量、形状、分布有着密切的关系。
2.陶瓷材料的分子结构特点
陶瓷晶体结构中,最重要的有氧化物结构与硅酸盐结构两类。大多数氧化物的结构是氧离子排列成简单立方、面心立方和密排六方的晶体结构,正离子位于其间隙中。它们主要是以离子键结合的晶体。图2-17所示为MgO与Al2O3的晶体结构。

图2-17 氧化物(MgO与Al2O3)晶体结构
硅酸盐结构也是陶瓷组织中的重要晶体相,它是以硅氧四面体[SiO4]为基本结构单元所组成的,为共价键结合,并带有离子键的特征。硅氧四面体结构如图2-18(a)、(b)所示,硅原子位于氧四面体正中央,其以共用顶点的O原子相互连接成岛状、链状、层状、骨架状等硅酸盐结构等大分子形态,如它们规则排列则形成晶体;否则,就为玻璃体,如图2-18(c)、(d)所示。如其间含有不同的粒子或离子,则又派生出不同性能的陶瓷或玻璃。
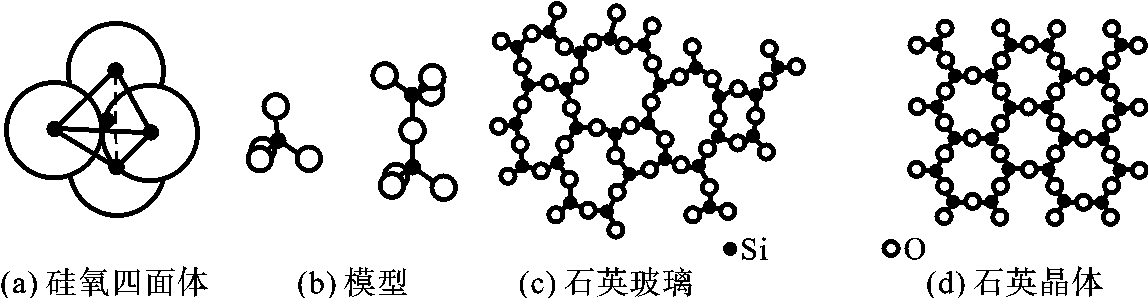
图2-18 硅酸盐结构示意图
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










