



第5节 膜转运载体蛋白病
一、肝豆状核变性
肝豆状核变性(hepatolenticular degeneration,HLD)(MIM 277900)是由于铜代谢障碍导致患者不能合成血浆铜蓝蛋白。本病由Wilson于1912年首次描述,故又称Wilson病(Wilson disease,WD)。主要病理改变为豆状核变性和肝硬化,临床上表现为进行性加剧的肢体震颤、肌强直、精神改变、肝硬化和角膜色素环(图9-23)形成。本病可分为两型:①晚发型,多在20~30岁间发病,病程进展缓慢;②少年型,多在7~15岁间发病,病程进展迅速。起病症状因人而异,大多数患者首先出现神经症状,少数先出现肝脏症状。
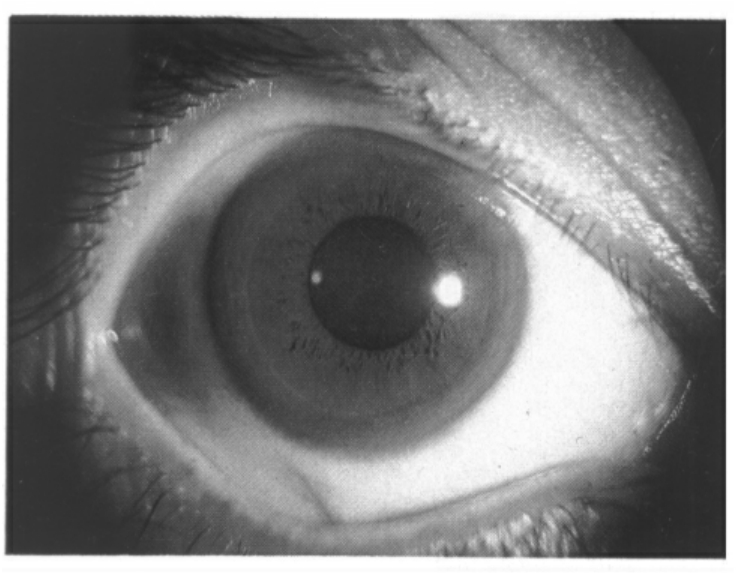
图9-23 肝豆状核变性(示角膜色素环)
本病为常染色体隐性遗传,群体发病率为1/100000~1/10000,铜代谢异常是该病发生的重要环节。导致铜代谢异常的机制曾先后提出五种学说:①胃肠对铜吸收过多;②铜蓝蛋白异常;③胆管排铜障碍;④溶酶体缺陷;⑤异常蛋白存在。异常蛋白存在学说是1953年由Uzman首次提出,在WD细胞内可能存在一种与铜离子有高度亲和力的异常蛋白,此蛋白与铜离子牢固结合,阻碍肝中铜蓝蛋白与铜的结合,使肝脏释放铜蓝蛋白减少,导致患者血清铜蓝蛋白降低。患者也有胆管排铜减少。
连锁分析表明,本病基因定位13q14.3,与酯酶D呈紧密连锁,共21个外显子,编码1411个氨基酸。我国学者的研究显示,第5外显子可能是中国人WD基因突变热点之一。该病的基因诊断可利用一些具有多态性的短串联重复序列(STR)在人类基因组中差异大、多态信息量高、与WD基因呈明显的连锁不平衡的特点,进行扩增片段长度多态性分析而确诊。另外,可以运用PCR-SSCP技术直接对WD基因中的某些突变热点所在的外显子进行检测,作为诊断指标。
本病若在肝硬化或神经系统症状出现前就进行治疗,并终生服药,可使患者维持正常的生活。
二、胱氨酸尿症
胱氨酸尿症(cystinuria,CSNU)(MIM220100)是遗传性近端肾小管上皮细胞和空肠黏膜对胱氨酸、赖氨酸、精氨酸、鸟氨酸的转运存在特异性缺陷,尿中此四种氨基酸排出过量。由于这四种氨基酸中赖氨酸、精氨酸和鸟氨酸均极易溶于水,而胱氨酸较不易溶于水,因而胱氨酸在尿中溶解度低,可结晶析出形成结石。
患者出生后即开始发病,但常于20~30岁时因突然结石发作才被确诊。患者通常体型矮小,智力低下,尿路结石反复发作,可导致尿路感染和绞痛。
本病为常染色体隐性遗传,至少涉及三个不同的异常等位基因,各以一种特异的方式影响主动转运过程。临床可分为以下三型。
Ⅰ型为常染色体隐性遗传,相关基因定位于2p16.3。患者肾小管和小肠的转运系统均异常,纯合体大量排出四种氨基酸,但杂合体和正常纯合体无氨基酸尿。
Ⅱ型为不完全隐性遗传,相关基因定位于2p16.3。纯合子患者四种氨基酸排出量均增加,而杂合体有中度氨基酸尿,尿中赖氨酸和胱氨酸增加,正常纯合体四种氨基酸排出量均正常。
Ⅲ型也为不完全隐性遗传,相关基因定位于19p13.1。纯合体患者尿中仅有轻度过量氨基酸,杂合体胱氨酸、赖氨酸排出量增加。
本病目前无根治办法,主要是通过控制饮食、药物治疗或手术等方式减少胱氨酸的排出和增加其溶解度,预防胱氨酸结石形成,减少或消除并发症。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










