



光催化氧化法是目前研究较多的一项高级氧化技术,一般可分为均相和非均相催化两种类型。均相光催化降解中较常见的是以Fe2+或Fe3+及H2O2为介质,通过photofenton反应产生使污染物得到降解,非均相光催化降解中较常见的是在污染体系中投加一定量的光敏半导体材料,同时结合一定量的光辐射,使光敏半导体在光的照射下激发产生电子 空穴对,吸附在半导体上的溶解氧、水分子等与电子空穴作用,产生·OH等氧化性极强的自由基,再通过与污染物之间的羟基加和、取代、电子转移等式污染物全部或接近全部矿化。
理论上来说,只要半导体具有合适的能带结构,使得其吸收的光能大于或等于能隙宽度,就能被激发产生光生电子和空穴对,该半导体就可能被用作光催化剂。目前广泛研究的半导体光催化剂大部分为金属的氧化物和硫化物,如Ti O2、Sn O2、Zn O、V2O5、Cd S、Zn S、Pb S和Mo Si2等。这些半导体中以Ti O2、Cd S和Zn O的催化活性最高,但Cd S和Zn O在光照射时不稳定,且因光阳极腐蚀产生Cd2+、Zn2+离子,对生物有毒性,对环境有害。此外,还有一类半导体材料(如Sr Ti O3),尽管与Ti O2具有同样的光催化性能和稳定性,但是,由于它们的吸收带隙均大于3.2e V,不利于可见光的直接吸收和利用,因而没有成为实用的光催化剂。
作为光催化剂Ti O2具有以下4个优点:
(1-)合适的半导体禁带宽度(3.2e V),可以用385nm以下的光源激发活化,通过改性有望直接利用太阳能来驱动光催化反应。
(2)光催化效率高,导带上的电子和价带上的空穴具有很强的氧化还原能力,可分解大部分有机污染物。
(3)化学稳定性好,具有很强的抗光腐蚀性。
(4)原料无毒且易得,成本较低。
1)Ti O2的结构与性质
Ti O2为白色粉末状多晶型化合物,俗称“钛白”,化学性质呈惰性,具有优异的颜料特性,对光的折射率高。Ti O2是常见的n型半导体,在自然界中有三种结晶形态:金红石型(四方晶系,P42/mnm),锐钛矿(四方晶系, I41/amd),板钛矿(无定型,Pbca)。板钛矿在自然界中很稀有,属斜方晶系,是不稳定的晶型,在650℃左右即转化为金红石型,因而工业价值不高;金红石和锐钛矿都属于四方晶系,但具有不同的晶格。两种晶格均由Ti O6八面体基元组成,不同的是八面体的畸变程度和八面体的连接方式不同,如图1-1所示。
图1-1 犜犻犗6结构单元的连接
(a)共边连接方式(锐钛矿型) (b)共顶点连接方式(金红石型)
金红石型的八面体由共顶点组成,不规则,微显斜方晶;锐钛矿由八面体共边组成,呈明显的斜方晶畸变,对称性低于前者。此外金红石Ti O2中的每个八面体与周围10个八面体相连,而锐钛矿Ti O2中每个八面体与周围8个八面体相连。锐钛矿Ti O2的Ti—Ti键长比金红石大,而Ti—O键长比金红石小。
组成金红石的Ti O6八面体是沿对角线方向拉长的八面体,因而Ti—O1键长比Ti—O2键长略长,但是O1—Ti—O2的键角没有变化,仍为90°。而在锐钛矿中,组成金红石的八面体的两个O1沿着四重轴的方向进一步发生畸变,因此锐钛矿八面体中的O1—Ti—O2不再是90°,金红石和锐钛矿的基本结构单元如图1-2所示。
图1-2 金红石和锐钛矿的犜犻犗6八面体结构
(a)金红石 (b)锐钛矿
晶体结构的差异使得不同晶型的Ti O2具有不同的质量密度和电子能带结构。锐钛矿型Ti O2的质量密度(3.894g/cm3)略小于金红石型Ti O2 (4.250g/cm3),带隙(3.3e V)略大于金红石Ti O2(3.1e V),由于金红石型的Ti O2的晶格较为紧密,有较大的稳定性和相对密度,因而具有较高的折射率和介电常数以及较低的热传导性。但金红石型Ti O2对O2的吸附能力较差,比表面积较小,光生电子和空穴容易复合,其光催化活性较低。金红石和锐钛矿型Ti O2物化性质比较如表1-2所示。
表1-2 金红石和锐钛矿型犜犻犗2的物化性质比较
续 表
Ti O2的不同晶型之间可发生转化。金红石相稳定,即使在高温下也不发生转化和分解,而锐钛矿和板钛矿相在加热过程中发生不可逆的放热反应,转变为金红石相。锐钛矿—金红石相转变为非平衡相变,相变发生在一定的温度范围(400~1000℃),而相变温度与杂质、颗粒大小、表面积等密切相关。另外,压力对Ti O2的多形态转变也有明显影响。高压下(大于2.6GPa),金红石相和锐钛矿相向高压相转变,压力增大至37.2GPa时相开始向另一高压相相转变。
2)Ti O2光催化反应原理
Ti O2是n型半导体,它之所以能够作为催化剂,与它自身的结构和特性有关。根据以能带为基础的电子理论,半导体的基本能带结构是由填满电子的价带(valenceband,VB)和空的导带(conductionband,CB)构成,价带和导带之间存在禁带。当用能量等于或大于禁带宽度(Eg)的光照射时,半导体价带上的电子可被激发跃迁到导带,同时在价带产生相应的空穴,这样就在半导体内部生成了电子(e-)空穴(h+)对。锐钛矿型Ti O2的禁带宽度是3.2e V (金红石型Ti O2的禁带宽度为3.4e V),当它吸收了波长等于或小于387.5nm的光子后,价带中的电子就会被激发到导带,形成带负电的高活性电子 ,同时在价带上产生带正电的空穴
,同时在价带上产生带正电的空穴 。与金属不同,半导体材料的能带之间缺少连续区域,电子 空穴对一般有皮秒级的寿命,足以使光生电子和光生空穴对经由禁带向来自溶液或气相的吸附在半导体表面的物种转移电荷。空穴可被半导体材料表面被吸附物质或溶剂中的电子,使原本不吸收光的物质被活化并被氧化,电子受体通过接受表面的电子而被还原。
。与金属不同,半导体材料的能带之间缺少连续区域,电子 空穴对一般有皮秒级的寿命,足以使光生电子和光生空穴对经由禁带向来自溶液或气相的吸附在半导体表面的物种转移电荷。空穴可被半导体材料表面被吸附物质或溶剂中的电子,使原本不吸收光的物质被活化并被氧化,电子受体通过接受表面的电子而被还原。
光诱发电子和空穴向吸附的有机/无机污染物或溶剂的转移,是电子和空穴向半导体表面迁移的结果。通常在表面上,半导体能够提供电子去还原电子接受体(在含有空气的水溶液中通常是氧),而空穴则能迁移到表面和供给电子的物种结合,从而使该物种氧化。在这个过程中,电子和空穴的电荷转移过程的速率和可能性取决于导带和价带各自的位置和被吸附物的氧化还原电位。下面以Ti O2在水溶液中的光催化反应为例,说明Ti O2光催化反应的步骤。
在半导体表面失去电子的主要为水分子、氢氧根离子和有机物,光生空穴能够同吸附在催化剂表面的OH-或H2O发生作用生成氧化能力极强的羟基自由基(·OH)。在p H=7时,羟基自由基的标准氧化还原电位为2.27V,是一种比臭氧还要强的氧化剂,能够无选择地氧化多种有机物并使之矿化,通常认为是光催化反应体系中主要活性氧化物种。光生电子的俘获剂主要是吸附在Ti O2表面的氧,既可以抑制电子与空穴的复合,也可以作为氧化剂,光生电子可以与之反应生成HO2·和 ·等活性氧类,同时也是表面羟基的另一个来源,半导体光催化反应机理如图1-3所示。
·等活性氧类,同时也是表面羟基的另一个来源,半导体光催化反应机理如图1-3所示。
图1-3 半导体光催化反应机理
光催化反应的基本反应式如下:当以波长小于387.5nm的光照射Ti O2半导体后,价带中的电子被激发到导带,产生光生电子空穴对,激发态的导带电子和价带空穴又能重新复合,使光能以热能或其他形式散掉。
Τi O2+hv → Τi O2+h++e- (1-1)
h++e → 复合+能量(hν或热能)(1-2)
当催化剂存在合适的俘获剂或表面缺陷态时,电子和空穴的重新复合得到抑制,在它们复合之前,就会在催化剂表面发生氧化 还原反应。价带空穴是良好的氧化剂,导带电子是良好的还原剂,在光催化半导体中,空穴具有更大的反应活性,一般与表面吸附的OH-或H2O发生反应形成具有强氧化性的羟基自由基(·OH)。
H2O+h+→·OH+H+ (1-3)
OH-+h →+ ·OH(1-4)
电子与表面吸附的氧分子反应,分子氧不仅参与还原反应,还是表面羟基自由基的另外一个来源:
O2+e-→· (1-5)
(1-5)
H2O+·O-→2 ·OOH+OH- (1-6)
2·OOH →O2+H2O2(1-7)
·OOH+H2O+e →- H2O2+OH- (1-8)
H2O2+e -→·OH+OH- (1-9)
反应中所生成的非常活泼的羟基自由基(·OH),超氧离子自由基(· )以及·OOH自由基等氧化性很强的活泼自由基,能够将各种有机物直接氧化成CO2和H2O等无机小分子,不产生中间产物。
)以及·OOH自由基等氧化性很强的活泼自由基,能够将各种有机物直接氧化成CO2和H2O等无机小分子,不产生中间产物。
从反应历程来看,通过光激发后,Ti O2产生高活性光生空穴和光生电子,形成氧化还原体系,经一系列反应产生大量高活性自由基,其中·OH是主要的自由基。氧化还原反应由两个半反应组成:氧化反应和还原反应,反应速率由速率较慢的半反应所决定。氧化物的电子还原反应(ms)大大慢于还原物的空穴氧化反应(1-00ns)。对于普通的光催化反应,光生电子e-向氧分子O2的转移是催化反应中的控速步骤,因为氧分子需要经过多个步骤才能变成具有强氧化性的羟基自由基(·OH)或超氧离子自由基(·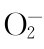 )等活性基团,从而将有机物彻底降解。
)等活性基团,从而将有机物彻底降解。
3)Ti O2光催化活性的影响因素
Ti O2的光催化活性与多个影响因素有关,不同晶型的Ti O2的光催化活性有很大区别,一般来说,锐钛矿型Ti O2的光催化活性高于金红石型Ti O2;在Ti O2不同的晶面上物质的光催化活性和选择性也有很大区别;而且实际晶体的表面即使在室温下也存在Ti2+、Ti3+等低价钛缺陷,可构成光催化反应的活性中心;另外,温度和溶液p H值的影响比较复杂,通常具有双重性;且Ti O2的表面结构对光催化活性也有较大影响。在本书中,主要讨论Ti O2的晶型和表面微观结构对光催化活性的影响。
(1-)Ti O2的晶型结构对光催化活性的影响。
Ti O2在自然界中以三种结晶形式存在:金红石型、锐钛矿型和板钛型。通常认为锐钛矿是活性最高的一种晶型,其次是金红石型,而板钛矿和无定型Ti O2没有明显的光催化活性。锐钛矿之所以表现出高的活性,其原因如下:
①锐钛矿的禁带宽度为3.2e V,而金红石的禁带宽度为3.0e V,较高的禁带宽度使得锐钛矿的电子空穴对具有更正或者更负的电位,因而具有更强的氧化能力。
②锐钛矿表面对H2O、O2及OH的吸附能力较强,而在光催化反应中,表面吸附能力对催化活性有很大的影响,较强的吸附能力使得其活性较高。
③锐钛矿晶粒在结晶过程中通常具有较小的尺寸及较大的比表面积,对光催化反应有利。
但是,简单地认为锐钛矿比金红石活性高是不严谨的,它们的活性并不仅仅由晶型所决定,而是受到其晶化过程的一些因素的影响。通常在同等条件下无定形Ti O2结晶成型时,金红石通常会形成大的晶粒,具有较差的吸附性能,由此导致金红石活性较低;但是,如果在结晶时能保持金红石与锐钛矿同样的晶粒尺寸及表面性质,则金红石也会表现出较高的活性。Lee等用脉冲激光照射锐钛矿Ti O2,由于晶体内部产生高温使得晶粒向金红石相转变,相转变的过程中使得比表面积和晶粒保持不变,研究发现这种方法制得的金红石型Ti O2表现出相当高的活性。另外Tsai等采用不同方法制备锐钛矿和金红石型Ti O2光催化降解含酚溶液,研究发现,Ti O2的活性不仅仅由晶型所决定,而是与制备方法及煅烧温度相关,在一定条件下,由于金红石型Ti O2表面存在大量的羟基,从而表现出很高的光催化活性。由此可见,Ti O2光催化剂的活性不仅仅由其晶型决定,无论是锐钛矿还是金红石相Ti O2,它们都可能具有较高的光催化活性,而其活性主要取决于晶粒表面的性质以及尺寸大小等因素。
值得注意的是,最近的研究表明:由锐钛矿和金红石以恰当比例组成的混晶通常比单一晶体的活性高。混合晶体表现出较高的活性是因为在结晶过程中,锐钛矿表面形成薄的金红石层,通过金红石层能有效地提高锐钛矿晶型中电子空穴分离效率,称为混晶效应。Bacsa等人用醇盐水解的方法制备Ti O2光催化剂发现,100%的锐钛矿与100%的金红石活性同样不高,而不同比例的两者混合体却表现出比纯的锐钛矿或金红石更高的活性,其中尤以30%金红石和70%锐钛矿组成的混合晶型的活性最高,由此可见,两种晶型的确具有一定的协同效应。
另外,Ohno等报道,Ti O2不同晶型的活性还与电子受体有关:以O2为电子受体时,锐钛矿的活性很高而金红石的活性很低;而以Fe3+为电子受体时,金红石表现出更高的活性。由于O2作为电子受体在光催化反应中对催化剂的材质非常敏感,而金红石的表面结构或其较低的导带能势可能是其对O2转输电子效率低的原因。而在大多数光催化反应的研究中,通常以O2作为电子受体,而金红石型以O2作为电子受体时表现出较低的活性,从而常常被认为活性较低。
(2)Ti O2的表面结构对光催化活性的影响。
一般来说,在以固体为催化剂的多相光催化反应中,催化反应过程发生在催化剂表面上。对于单纯的Ti O2光催化剂,影响光催化活性的表面性质有:①表面积,尤其是充分接受光照的表面积;
②表面对光子的吸收能力;
③表面对光生电子和空穴的捕获并使其有效地分离的能力;
④电荷在表面向底物转移的能力。
表面对光子的吸收能力常难以与光催化活性直接相关联,因为表面的改变通常引起其他结构的变化,直接牵连多个因素的变动,使得其表面结构成为Ti O2光催化活性研究中的重点。
4)Ti O2光催化剂的制备
Ti O2光催化剂主要有两种形式,即纳米Ti O2粉体和Ti O2薄膜。
(1-)纳米Ti O2粉体的制备方法。
目前,制备Ti O2粉体的方法可分为物理制备法和化学合成法两大类。
物理制备法是指借助物理加工方法得到纳米尺度结构的Ti O2的方法,常用技术有离子溅射法、射频磁控溅射法、机械研磨法等。Anpo等利用射频磁控溅射的方法在石英基片上得到尺度可控的纳米Ti O2薄膜,研究发现,在高温下(如873K)用该方法制得的Ti O2膜出现严重的氧原子流失,钛氧原子比在膜表面约为Ti/O=1.93,Ti O2膜的吸收边缘也从387nm延伸到了550nm以上。
化学合成法可归纳为气相法和液相法两大类。目前实验室和工业上广泛采用液相法制备纳米粉体Ti O2,液相法具有低温合成、设备简单、易操作、成本低等优点。气相法是利用气态物质在固体表面进行化学反应,生成固态沉积物的过程。气相法包括气相氧化法、气相水解法、化学气相沉积法等。利用气相法制得的Ti O2纳米粉体具有纯度高、粒度小、单分散性好、团聚小等优点,但设备复杂、能耗大、成本高。
①液相法合成Ti O2。
液相法合成Ti O2主要有液相沉淀法、溶胶 凝胶法、醇盐水解法、微乳液法及水热法等。
a.液相沉淀法:
液相沉淀法合成纳米Ti O2粉体,一般以Ti Cl4或Ti(SO4)2等无机钛盐为原料,原料便宜易得,是最经济的制备方法。通常采用的工艺路线是将氨水、(NH4)2CO3或Na OH等碱类物质加入到钛盐溶液中,生成无定形的Ti(OH)4;将生成的沉淀物过滤、洗涤、干燥后,经600℃左右煅烧得锐钛矿型、800℃以上得到金红石型纳米Ti O2粉体。
近年来,在缩短制备流程,改进沉淀工艺方面进行了不少研究。赵敬哲等引入胶溶作用,提出液相一步合成金红石型超细Ti O2粉体的工艺。该工艺是将洗净的无定形Ti(OH)4沉淀重新分散于2mol/L的HNO3溶液中,在80℃加热回流胶溶2h后,沉淀经离心、干燥,得到粒径为数十纳米的金红石型Ti O2粉体。这种工艺省去耗能多的高温煅烧过程,同时避免了在此过程因烧结而引起的纳米粒子之间的硬团聚。
Hee Dong Nam等提出一种不需要煅烧工序的更为简便的直接升温水解工艺。该工艺不加入碱性物质沉淀剂,只将Ti OCl2水溶液升温,使其发生水解,在液相中直接生成晶型沉淀,将沉淀产物干燥后,即得锐钛矿型或者金红石型纳米Ti O2粉体。通过改变原料液浓度、水解温度、反应时间等工艺条件,可以控制产物晶型、粒度大小等特性,水解温度在65℃以下可得到金红石型沉淀,在100℃左右可得到锐钛矿型沉淀。
采用液相沉淀法合成纳米Ti O2粉体,必须通过液固分离才能得到沉淀物,由于引入大量SO2-4或Cl-等无机离子,需要反复洗涤以除去这些离子,因而工艺流程较长、废液多、产物损失较大,而且因为完全洗净无机离子较困难,因而制得的Ti O2粉体纯度不高,难以适用对纳米Ti O2纯度要求高的应用领域。
b.溶胶凝胶法:
溶胶凝胶法合成纳米Ti O2粉体一般以钛醇盐Ti(OR)4(R=C2H5,—C3H7,—C4H9)为原料,其主要步骤是:钛醇盐溶于溶剂中形成均相溶液,以保证钛醇盐的水解反应在分子均匀的水平上进行,由于钛醇盐在水中的溶解度不大,一般选用醇(乙醇、丙醇、丁醇等)作为溶剂;钛醇盐与水发生水解反应,同时发生失水和失醇缩聚反应,生成物聚集成1nm左右的粒子并形成溶胶;经陈化,溶胶形成三维网络而形成凝胶;干燥凝胶以除去残余水分、有机基团和有机溶剂,得到干凝胶;干凝胶研磨后煅烧,除去化学吸附的羟基和烷基团,以及物理吸附的有机溶剂和水,得到纳米Ti O2粉体。因为钛醇盐的水解活性很高,所以需加抑制剂来减缓其水解速度。常用的抑制剂有盐酸、氨水、硝酸等。溶胶 凝胶法制备Ti O2工艺流程如图1-4所示。
图1-4 溶胶凝胶法制备TiO2工艺流程
溶胶凝胶法的反应过程如下。
水解反应:
Ti(OR)4+nH2O→Ti(OR (OH)n+nROH(1-10)
(OH)n+nROH(1-10)
缩聚反应:
2Ti OH→Ti OTi+H2O(1-11)
Ti OR+ HOTi→ Τi OTi+ROH(1-12)
溶胶化反应:
Ti(OR)4+mR′OH→Ti(OR)(4-m)(OR′)m+mROH (1-13)
采用溶胶凝胶工艺合成的纳米Ti O2粉体,具有反应温度低(通常在常温下进行),设备简单,工艺可控可调,过程重复性好等特点,与沉淀法相比,溶胶凝胶法不需要过滤洗涤,避免产生大量废液,同时,因凝胶的生成,凝胶中颗粒间结构的固定化,还可以有效抑制颗粒的生长和团聚过程,得到粒度细且单分散性好的粉体。
c.醇盐水解沉淀法:
醇盐水解沉淀法与上述的溶胶凝胶法一样,也是利用钛醇盐的水解和缩聚反应,但设计的工艺过程不同,此法是通过醇盐水解、均相成核与生长等过程在液相中生成沉淀产物,再经过液固分离、干燥和煅烧等工序,制备Ti O2粉体。醇盐水解沉淀法合成Ti O2粉体的工艺流程如图1-5所示。
图1-5 醇盐水解法合成犜犻犗2的工艺流程
醇盐水解沉淀法的反应对象主要是水,不会引入杂质,所以能制备高纯度的Ti O2粉体;水解反应一般在常温下进行,设备简单,能耗少。然而,因为需要大量的有机溶剂来控制水解速度,致使成本较高,如果能实现有机溶剂的回收和循环使用,则可有效地降低成本。
d.微乳液法:
微乳液法是近年来刚开始被研究和应用的方法。微乳液是由表面活性剂、助表面活性剂(通常为醇类)、油(通常为碳氢化合物)和水(或电解质溶液)组成的透明的、各向同性的热力学稳定体系,可分成O/W型微乳液和W/O型微乳液。W/O型微乳液的微观结构由油连续相、水核及表面活性剂与助表面活性剂组成的界面三相组成,其中,水核可以看作一个“微型反应器”,其大小可控制在几到几十纳米之间,彼此分离,是理想的反应介质。
当微乳液体系确定后,超细粉的制备是通过混合两种含有不同反应物的微乳液实现的。其反应机理是,当两种微乳液混合后,由于胶团颗粒的碰撞,发生了水核内物质的相互交换和传递,这种交换非常快。化学反应就在水核内进行,因而粒子的大小可以控制。一旦水核内粒子长到一定尺寸,表面活性剂分子将附在粒子的表面,使粒子稳定并防止其进一步长大。微乳液中反应完成后,通过超离心或加入水和丙酮混合物的方法,使超细颗粒与微乳液分离,再用有机溶剂清洗,以去除附在粒子表面的油和表面活性剂,最后在一定温度下干燥,煅烧后得到超细粉。
微乳液的结构从根本上限制了颗粒的生长,使超细粉末的制备变得容易实现。微乳液法不需加热、设备简单、操作容易且制得的颗粒大小可控。但是,由于使用了大量的表面活性剂,这些有机物很难从最终获得的粒子表面去除,对制得粉末的纯度有影响。
e.水热法:
水热法是在特制的密闭反应容器(高压釜)里,采用水溶液作为反应介质,通过对反应容器加热,创造一个高温、高压反应环境,使得通常难溶或不溶的物质溶解并且重结晶,通常采用固体粉末或新配制的凝胶作为前驱体。水热法制备纳米Ti O2粉体,第一步是制备钛的氢氧化物凝胶,反应体系有四氯化钛+氨水和钛醇盐+水。第二步是将凝胶转入高压釜内,升温(通常的温度为120~250℃),造成高温、高压的环境,使难溶或不溶的物质溶解并且重结晶,生成纳米Ti O2粉体。
水热法能直接制得结晶良好的粉体,省去高温灼烧处理工艺,避免了在此过程中可能形成的粉体硬团聚,而且通过改变工艺条件,可以控制粉体粒径、晶型等特性。同时,因经过重结晶,所以制得的粉体纯度高。但由于该工艺需要在高温高压的环境中进行,因而对设备要求高,操作复杂,能耗较大。
②气相法合成Ti O2。
气相法合成Ti O2主要有气相水解法、气相氧化法、化学气相沉积法和蒸发凝聚法等。
a.气相水解法:
气相水解法又称气溶胶法,将Ti Cl4从气体导入高温氢氧焰中(700~1000℃)进行气相水解。该方法最早由德国德固萨公司(Degussa)开发成功, P25纳米Ti O2粉体就是用该方法生产的,其中约含锐钛矿型Ti O270%,金红石型Ti O230%,平均粒径在30nm左右,比表面积为50m2/g。气相水解法不直接采用水蒸气水解,而是靠氢氧焰燃烧产生的水蒸气水解,反应温度达到1800℃左右,超过Ti O2的熔点,生成的Ti O2颗粒呈气溶胶状态,因此又称气溶胶法。气相水解法的反应式如下。
氢燃烧反应:
2H2(g)+O2→2H2O(g)(1-14)
Ti Cl4水解反应:
Ti Cl4(g)+2H2O(g)→Τi O2(s)+4HCl(g)(1-15)
总反应:
Ti Cl4(g)+2H2O(g)+O2(g)→Τi O2(s)+4HCl(g) (1-16)
气相水解法可通过调节过程中温度、料比、流量、反应时间等参数控制Ti O2的粒径大小和晶型等特性,且制得的Ti O2粉体纯度高、粒径分布窄、表面光滑、无孔、分散性好和团聚程度小。
b.气相氧化法:
气相氧化法是通过将Ti Cl4在高温下氧化来制备Ti O2。在反应初期, Ti Cl4和O2发生均相化学反应,生成Ti O2的前驱体分子,通过成核形成Ti O2的分子簇或粒子。由于非均相成核比均相成核在热力学上更容易,随着反应的进行,Ti Cl4在Ti O2粒子表面吸附并进行非均相反应,使粒子变大。在气相氧化法中,反应温度、停留时间以及冷却速度都将影响气相氧化法得到的Ti O2的粒子形态。
Ti Cl4气相氧化法具有节能、环保、成本低和自动化程度高的优点,可以分别制备出锐钛矿型、混晶型和金红石型纳米Ti O2粉体,在气相法中最具有竞争力。但由于气相氧化法是一个复杂的过程,一方面在发生化学反应和成核、生长的同时,Ti O2分子、分子簇和粒子之间会发生碰撞、凝并成团聚体,在高温下还会发生烧结和晶型转化等固相反应,而且各过程并非是简单的并联和串联过程;另一方面,由于反应器涉及复杂的流动、传递、混合等工程问题,也会影响纳米Ti O2的结构和性能。
c.蒸发凝聚法:
利用高频等离子技术对工业的Ti O2粗品进行加热,使其汽化蒸发,再急速冷却可得到纳米级的Ti O2。
(2)Ti O2薄膜的制备方法。
①物理气相沉积法。
物理气相沉积(PVD)是薄膜制备的常用技术,与化学气相沉积法(沉积粒子来源于化合物的气相分解反应)相比,PVD的沉积温度较低,不易引起基底的变形与开裂以及薄膜性能的下降。Ti O2薄膜可以通过电子束蒸发、活化反应蒸发、离子束溅射、离子束团束(ICB)技术、直流/交流反应磁控溅射、高频反应溅射、分子束外延等物理气相沉积的方法制备。其中反应磁控溅射金属Ti靶的方法,能制备出具有较高折射率的高质量的Ti O2薄膜,其制备工艺稳定、制备条件易于控制,能够在建筑玻璃等大规模生产中得到应用。
Takeda等用金属Ti靶通过直流磁控溅射技术在有Si O2阻挡层的玻璃基板上制备光催化活性的Ti O2薄膜。薄膜可在大面积内保持厚度均匀,在可见光区透射率约为80%。在紫外光照射下,Ti O2薄膜对乙醛的分解能力与溶胶凝胶方法制备的薄膜基本一致,但溅射的Ti O2薄膜具有更好的机械强度。
应用脉冲激光沉积(PLD)技术也是制备Ti O2薄膜的有效方法之一。秦启宗等在含O3的O2气氛中,用355nm的脉冲激光烧蚀金属钛靶在ITO (In2O3Sn O2)玻璃基片上反应沉积Ce O2/Ti O2薄膜,研究了Ti O2薄膜的电学性能和介电性能以及Ti O2薄膜电极的电化学嵌锂过程。以活化反应蒸发(activereactivevaporization)技术制备Ti O2光催化薄膜。在反应装置中,通入0.1~0.2Pa的O2气氛,将热阴极加热并加上高压直流电源使气体处于辉光放电状态,用钨蒸发器加热钛丝,蒸发的钛丝在载体上形成Ti O2薄膜。然后将Ti O2膜置于空气中在400℃下退火,根据镀膜时间及退火时间的不同可得到性能不同的催化剂膜。
物理气相沉积方法制备的薄膜均匀,厚度易控制,是一种工业上广泛应用的制膜方法,但所需的设备价格较昂贵。
②溶胶凝胶法。
溶胶凝胶法也是制备薄膜最常用且最有效的方法之一。溶胶凝胶法技术具有纯度高、均匀性好、合成温度低(甚至可在室温下进行)、化学计量比及反应条件易于控制等优点,特别是制备工艺过程相对简单,无须特殊贵重的仪器。
溶胶凝胶法制备薄膜时,先将金属有机醇盐或无机盐进行水解、聚合,形成金属盐溶液或溶胶,然后用提拉法、旋涂法或喷涂法等将溶胶/溶液均匀涂覆于基板上形成多孔、疏松的干凝胶膜,最后再进行干燥、固化及热处理即可形成致密的薄膜。用溶胶凝胶技术制备Ti O2薄膜常用的含钛的前驱体主要是钛醇盐,如钛酸四丁酯Ti(O Bu)4、Ti Cl4、Ti Cl3和Ti(SO4)2等,催化剂常用无机酸,如硝酸、盐酸。先将钛酸四丁酯与有机溶剂如异丙醇或乙醇等混合均匀,在不断搅拌下将混合溶液滴加到含适量酸的水中,形成透明的Ti O2的胶体,其反应过程如表达式(1-17)~式(1-18)所示:
水解: nM(OR)4+4nH2→O nM(OH)4+4nROH(1-17)
缩合: nM(OH) →4 nMO2+2nH2O(1-18)
总反应: nM(OR)4+2nH2→O nMO2+4nROH(1-19)
应用溶胶凝胶方法制备的Ti O2薄膜经一定温度焙烧后,溶胶凝胶中的有机物基本挥发和分解,薄膜中的Ti O2粒子呈纳米晶网络海绵状,具有很大的表面积和粗糙度,易吸附其他如染料等活性物质,使对Ti O2薄膜进行敏化时有较高的效率,这是其他制膜方法所不能比拟的。用溶胶凝胶法还可在室温下制备具有良好光催化性能和超亲水性能的Ti O2薄膜,并且薄膜具有较好的附着力,这是用其他方法难以实现的。
除以上方法外,还可采用液相沉积法、喷雾热分解法等方法制备Ti O2薄膜。
5)Ti O2光催化剂降解污水处理
Ti O2在废水处理中的应用主要可以分为处理废水中的有机污染物和无机污染物两个方面。
(1-)Ti O2光催化降解废水中的有机污染物。
Ti O2能有效地将废水中的有机物降解为H2O、CO2、P 、S
、S 、N
、N 卤素离子等无机小分子,达到完全无机化的目的。染料废水、农药废水、表面活性剂、氯代物、氟利昂、含油废水等都可以被Ti O2催化降解。Blake在一篇综述中详细罗列了300多种可被光催化的有机物。美国环保局公布的114种有机物均被证实可通过光催化氧化降解矿化。可采用n Ti O2光催化处理的有机废水及有机物的种类如下:
卤素离子等无机小分子,达到完全无机化的目的。染料废水、农药废水、表面活性剂、氯代物、氟利昂、含油废水等都可以被Ti O2催化降解。Blake在一篇综述中详细罗列了300多种可被光催化的有机物。美国环保局公布的114种有机物均被证实可通过光催化氧化降解矿化。可采用n Ti O2光催化处理的有机废水及有机物的种类如下:
染料废水:甲基橙、甲基蓝、罗丹明 6G、罗丹明B、水杨酸、羟基偶氮苯、水杨酸、分散大红、含磺酸基的极性偶氮染料等。
农药废水:除草剂、有机磷农药、三氯苯氧乙酸、2,4,5 三氯苯酚, DDVP、DTHP、DDT等。
表面活性基:十二磺基苯磺酸钠、氯化苄基十二磺基二甲基胺、壬基聚氧乙烯苯、乙氧基烷基苯酚等。
氯代物:三氯乙烯、三氯代苯、三氯甲烷、四氯化碳、4氯苯酚、2氯代二苯并二!英、2,7二氯代二苯并二!英、多氯代二苯并二!英、四氯联苯、氟利昂、五氟苯酚、氟代烯烃、氟代芳烃等。
油类:水面漂浮油类及有机污染物。
(2)Ti O2用于处理无机废水。
许多无机物在Ti O2表面也具有光化学活性,Miyaka等早在1977年就用Ti O2悬浮粉末光解Cr2O 还原为Cr3+的研究。利用二氧化钛催化剂的强氧化还原能力,还可以将污水中汞、铬、铅以及氧化物等降解为无毒物质。Frank等研究了以Ti O2等为催化剂将CN-氧化为OCN-,再进一步反应生成CO2、N2和NO
还原为Cr3+的研究。利用二氧化钛催化剂的强氧化还原能力,还可以将污水中汞、铬、铅以及氧化物等降解为无毒物质。Frank等研究了以Ti O2等为催化剂将CN-氧化为OCN-,再进一步反应生成CO2、N2和NO 的过程。Serpone等报道了用Ti O2光催化法从Au(CN
的过程。Serpone等报道了用Ti O2光催化法从Au(CN 中还原Au,同时氧化CN-为NH3和CO2的过程,指出该法用于电镀工业废水的处理,不仅能还原镀液中的贵金属,而且还能消除镀液中氰化物对环境的污染,是一种有实用价值的处理方法。
中还原Au,同时氧化CN-为NH3和CO2的过程,指出该法用于电镀工业废水的处理,不仅能还原镀液中的贵金属,而且还能消除镀液中氰化物对环境的污染,是一种有实用价值的处理方法。
由于早期的悬浮型Ti O2体系中Ti O2粉末是以悬浮态存在于水溶液中,所以存在催化剂难以回收,活性成分损失较大、在水溶液中易于凝聚等问题而很难成为一项实用的水处理技术。近年来为了开发高效实用的光化学反应器,固定相光催化的研究逐步活跃起来。固定相光催化研究的焦点是负载型光催化剂的制备。
光催化剂载体的主要作用有以下几点:
①用载体将Ti O2固定,可防止Ti O2粉末粒子的流失并且易于回收利用,克服了悬浮相Ti O2的缺点;
②将Ti O2负载于载体表面,能够避免悬浮相中Ti O2颗粒的团聚,增加Ti O2的比表面积,提高Ti O2的利用率;
③有些载体可成为电子的俘获中心,有利于电子空穴对的分离,有些载体具有吸附性能,可增加对反应物的吸附,从而提高Ti O2的光催化活性;
④将Ti O2制成薄膜后,不存在催化剂粒子间的遮蔽问题,受到光照射的催化剂粒子数目增加,提高光源的利用率;
⑤用载体将催化剂固定,便于对催化剂进行表面修饰并制成各种形状的反应器。
因为Ti O2在紫外光照下能催化氧化有机物,故所选用载体多为无机或惰性有机材料,以硅酸类为主,其次还有金属、活性炭、沸石、高分子聚合物等。载体材料之间的不同的物理、化学性质,决定了负载Ti O2的特点。
①玻璃类载体具有较好的透光性,相应地对光的利用率高,但玻璃表面比较光滑,使得附着性能相对较差,此外由于Na+,Si4+在负载过程中热处理时可从载体表面迁移到Ti O2层,破坏Ti O2的晶格结构,成为电子空穴复合中心,从而降低Ti O2光催化活性。
②金属类载体可塑性好,但价格比较昂贵,而且Fe,Cr等金属离子在热处理时会进入Ti O2层,破坏Ti O2晶格降低催化活性,限制了金属类载体的使用。
③吸附剂类载体主要有硅胶、活性炭、沸石等,本身具有较大的比表面积,且具有吸附性能,使得污染物在溶液中与催化剂能够较充分的接触,可将有机物吸附到Ti O2粒子周围,增加局部浓度,避免反应的中间产物挥发或游离,加快光催化反应速度。利用这类载体存在的主要问题是Ti O2催化剂与载体之间的结合牢固程度需要进一步提高。
催化剂载体的选择需综合考虑多方面因素,如光利用效率、光催化活性、催化剂负载的牢固性、使用寿命、成本价格等。
Ti O2光催化技术应用于废水处理显示出了诱人的发展前景,它以操作简单、条件温和以及低能耗、不产生二次污染等突出特点,在水污染治理中有着其他方法不可替代的优势。尤其是对于一些其他方法难以降解有机物,如化学性质很稳定的苯,其降解效果是其他方法难以比拟的。但是,纳米Ti O2的光催化技术无论在理论基础研究还是在应用研究都还不成熟,离大规模生产还有一段距离,还有许多亟待解决的问题。
①光催化反应量子产率低(4%),最高不超过10%,难以处理量大且浓度高的工业废气和废水。太阳能利用率低,Ti O2光催化剂只能吸收利用太阳光中的紫外线部分,并不能充分利用太阳光的可见光部分。研发可见光诱导的新型高效光催化剂实现有机物在可见光下的分解,突破现有二氧化钛光催化剂量子效率和可见光利用率低的限制。迄今的光催化研究基本上利用的都是紫外光源,在实用中受到了一定的限制。开发新型或改性现有的催化剂,使它的光响应波长能移至可见光区从而直接利用太阳光作辐射源,将会使这项技术向实用化迈进一大步。
②提高催化剂的利用效率,包括充分利用催化剂表面积和采用辅助方法提高已有催化剂的活性。有人采用电化学辅助光催化的方法,将Ti O2膜固定在光电化学电池的阳极板上,光催化进行的同时在电极上加压,使光生电子刚产生就马上转移到电池阳极上,减少与空穴的复合机会,以此提高光催化效率。也有人采用微波辅助使光催化剂性能明显改善。
③光催化剂的负载技术,难以在保持了高的催化活性又满足特定材料的理化性能要求的前提下,在不同材料表面均匀、牢固地负载催化剂,使得催化剂不易分离再生。光反应器内负载在吸附载体上的催化剂流化操作,可以使催化剂的表面积充分受光,提高表面积利用率。
由于可以使用外加电场的方法实现光生电子与空穴的分离,从而提高Ti O2光催化活性,所以依据导电性能可将常用的载体分为导电性载体和绝缘性载体。玻璃、陶瓷、空心微珠、纤维网、多孔Al2O3等属于绝缘性载体,不能使用外加电场的形式进一步提高其光催化活性。导电玻璃、钢铁、铝、钛等金属是导电性载体,以它们作为载体制备的负载型Ti O2可以作为光电催化电极,使用外加电场的形式进一步提高其光催化活性。在这些导电负载体中,由于金属钛与Ti O2有很好的相容性,负载的Ti O2与基体结合力强,不易失活。所以以钛板为基体的负载型Ti O2电极倍受关注。目前研究最多的就是使用阳极氧化的方法在钛板表面制备一层Ti O2纳米管阵列。另外还有几种不同形貌的Ti O2膜。为了提高Ti O2膜的光催化活性,一般的方法是制备多孔膜增大其比表面积的方法。Ti O2膜形貌的改变不但可以改变其比表面积,而且改变其对光的吸收率和与水的接触面积,这些都与其光催化活性密切相关。既然Ti O2薄膜的表面形貌影响着其表面积、与水的接触面积和吸光度,进而影响着其光催化活性。那么改变、控制其表面形貌就对提高其光催化活性有着极大的意义。
(3)Ti O2膜结构。
Ti O2膜的表面微结构不但影响着Ti O2膜的比表面积,更为重要的是影响其有效比表面积以及表面附近水溶液与主体溶液的传质过程。另外表面微结构对Ti O2膜的吸光率有着极为重要的影响,以下为几种典型的Ti O2膜结构。
①平面膜结构。
最初负载型Ti O2膜是使用溶胶凝胶方法在平面载体如玻璃,其模型结构如图1-6所示。
图1-6 平面膜结构模型
平面膜比表面积为
式中:S为比表面积 为平面膜的上表面积;mTi O2为Ti O2膜的质量
为平面膜的上表面积;mTi O2为Ti O2膜的质量 为Ti O2膜的体积
为Ti O2膜的体积 为Ti O2的密度;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;d为Ti O2膜的厚度。
为Ti O2的密度;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;d为Ti O2膜的厚度。
从式(1-20)可以看出,平面膜的比表面积与膜厚度成反比。锐钛矿相Ti O2的密度为3.9g/cm3。一般使用溶胶凝胶法在玻璃表面提拉出的Ti O2膜层厚度为0.2μm。其比表面积约为1.28m2/g。
平面Ti O2膜比表面积与厚度关系如图1- 7所示,可以看出,膜厚度越薄,其比表面积越大。但是这种以减小膜层厚度、降低Ti O2质量而增大比表面积的方法并没有使得实际的表面积增加。无论厚度如何改变,其表面积没有改变。由此可见通常意义上的比表面积不能适用于这种薄膜模型。为此本书中对薄膜的比表面积定义为单位沉积面积上的膜的表面积。
图1-7 平面犜犻犗2膜比表面积与厚度之间的关系
式中:S膜为膜的比表面积;S表为膜的表面积;S沉积为沉积薄膜所占面积。
式(1-21)所定义的膜的比表面积是指膜的表面积与沉积膜所占的平面的面积之比,它是一个比值,是没有量纲的量,没有单位。以后如没有特别说明,膜的比表面积就指式(1-21)所定义的膜的比表面积。这样,平面膜比表面积就可用式(1-22)计算:式中:S膜为平面膜的比表面积;S表为平面膜的上表面积;S沉积为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽。
平面膜比表面积就为1。
②多孔膜结构。
由于大的比表面积有利于提高Ti O2膜光电催化活性,所以也有使用在溶胶中添加有机造孔剂、使用微弧氧化等方法制备多孔Ti O2膜。其模型结构如图1-8所示。
图1-8 多孔膜结构模型
图1--8中,为便于计算,假设多孔膜的孔为均一的,孔半径为R,孔深为d,孔密度为ε,则多孔膜的比表面积为
式中:S膜为多孔膜的比表面积;S表为多孔膜的表面积;S沉积为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为多孔膜的孔密度;R为多孔膜的孔半径;d为多孔膜的孔深。
从式(1-23)可以看出,多孔膜的比表面积主要与多孔膜的孔密度、孔半径以及膜厚度有关。孔密度越大,孔半径越大,孔越深其比表面积也就越大。
图1-9 孔呈密排六方排列
在半径一定情况下,孔密度最大值为密排六方时的孔密度,如图1--9所示。
图中,孔半径为R;犗犃=2犃犅;此时孔密度为
式中:ε为多孔膜的孔密度;S为每个孔所占据的面积;R为多孔膜的孔半径。
孔径与孔密度之间的关系图如图1-10所示。
图1-10 密排时孔密度与孔半径之间的关系
结合式(1-23)和式(1-24)可得
孔径和孔深与比表面积之间的关系如图1-11所示。
图1-11 孔径和孔深与比表面积之间的关系
从图1-11可以看出当孔径比较大的时候,孔深对比表面积的影响比较大。而当孔径比较小的时候,孔径对比表面积的影响比较大。
③纳米管膜结构。
2001年Grimes等使用阳极氧化的方法,首次在钛表面制备出具有纳米管阵列的Ti O2膜。其模型结构如图1-12所示。
图1-12 纳米管膜结构模型
如图1-12所示,为便于计算假设纳米管是均一的,管外径为R,内径为狉,深度为d,纳米管膜的比表面积为
式中:S膜为纳米管膜的比表面积;S表为纳米管膜的表面积;S沉积为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为纳米管的密度;R为纳米管的外半径;狉为纳米管的内半径;d为纳米管的深度。
从式(1-26)可以看出,纳米管膜的比表面积主要与纳米管的密度、外半径、内半径以及深度有关。比表面积随着纳米管的密度、外径、内径、和深度的增大而增大。
与多孔膜结构类似,当纳米管以密排六方形式排列时其密度最大。将式(1-24)代入式(1-26)得
设犽为纳米管内外径之比:
k=r/R (1-28)
则有
图1-13表示当纳米管深度为1000nm时,纳米管内外径与比表面积之间的关系。
图1-13 纳米管内外径与比表面积之间的关系
从图1-13可以看出纳米管膜的比表面积随着纳米管外径的减小、内外径比值的增大而增大。
图1-14表示纳米管内外径比值为0.9时纳米管深度与比表面积之间的关系。
图1-14 纳米管深度与比表面积之间的关系
纳米管深度对其比表面积影响比较大,纳米管比表面积随深度的增加而增大。
④尖劈结构膜结构。
考虑到Ti O2膜对光吸收问题,为了最大限度提高Ti O2膜对光的吸收,减小反射带来的损失,本书中设计了有几种不同结构单元组成的光陷阱结构Ti O2膜。它们的结构如图1-15所示。
各种尖劈结构膜的比表面积计算公式可以分别用以下几个公式计算:
a.尖劈模型(a)是由圆锥单元按四方阵列组合而成。设圆锥半径为R,高度为d,则其比表面积为(1-30)式中: 为膜的比表面积;S表为四方阵列圆锥尖劈膜的表面积
为膜的比表面积;S表为四方阵列圆锥尖劈膜的表面积 为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为圆锥的密度;R为圆锥的半径;d为圆锥的高度。
为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为圆锥的密度;R为圆锥的半径;d为圆锥的高度。
图1-15 不同单元结构的尖劈膜结构模型
(a)四方圆锥阵列尖劈结构模型 (b)密排圆锥阵列尖劈结构模型
(c)密排六棱锥尖劈结构模型 (d)密排四棱锥尖劈结构模型 (e)密排三棱锥尖劈结构模型
图1-16 圆锥呈四方排列
当圆锥按四方排列时如图1- 16所示,其密度为
ε= =
= (1-31)式中:ε为圆锥密度;S为每个圆锥所占据的面积;R为圆锥半径。
(1-31)式中:ε为圆锥密度;S为每个圆锥所占据的面积;R为圆锥半径。
-圆锥半径与圆锥密度之间的关系图如图1--17所示。
图1--17 四方排列时圆锥密度与半径之间的关系
将式(1-31)代入式(1-30)得
式中: 为膜的比表面积;ε为圆锥的密度;R为圆锥的半径;d为圆锥的高度。
为膜的比表面积;ε为圆锥的密度;R为圆锥的半径;d为圆锥的高度。
图1-18表示四方排列时圆锥半径和高度与比表面积之间的关系。
图1--18 四方排列时圆锥半径和高度与比表面积之间的关系
从图1--18可以看出,只有当圆锥半径很小、高度很高时对提高表面积作用比较明显。
b.尖劈模型(b)是由圆锥单元按密排六方阵列组合而成。其比表面积可由式(1-30)计算,圆锥密度可由式(1-24)得出。将式(1-24)代入式(1-30)得
式中 为膜的比表面积;ε为圆锥的密度;R为圆锥的半径;d为圆锥的高度。
为膜的比表面积;ε为圆锥的密度;R为圆锥的半径;d为圆锥的高度。
图1-19表示六方密排时圆锥半径和高度与比表面积之间的关系。
图1--19 六方密排时圆锥半径和高度与比表面积之间的关系
从图1--19可以看出,只有当圆锥半径很小、高度很高时对提高表面积作用比较明显。
c.尖劈模型(c)是由正六棱锥单元按密排六方排列而成。设其底面边长为犮,高为d,则其比表面积为
式中 为膜的比表面积
为膜的比表面积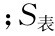 为尖劈膜的上表面积
为尖劈膜的上表面积 为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为六棱锥的密度;犮为六棱锥的底面边长;d为六棱锥的高度。
为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为六棱锥的密度;犮为六棱锥的底面边长;d为六棱锥的高度。
六棱锥密度为
式中:ε为六棱锥密度;S为每个六棱锥所占据的面积;犮为正六棱锥底面边长。
将式(1-35)代入式(1-34)得
式中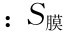 为膜的比表面积;ε为六棱锥的密度;犮为六棱锥的底面边长;d为六棱锥的高度。
为膜的比表面积;ε为六棱锥的密度;犮为六棱锥的底面边长;d为六棱锥的高度。
图1-20表示六方密排时正六棱锥高度和底面边长与比表面积之间的关系。
图1-20 六方密排时正六棱锥半径和高度与比表面积之间的关系
d.尖劈模型(d)是由正四棱锥以四方密排组成。设其底面边长为犮,高为d,则其比表面积为
式中 为膜的比表面积
为膜的比表面积 为尖劈膜的表面积
为尖劈膜的表面积 沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为四棱锥的密度;犮为四棱锥的底面边长;d为四棱锥的高度。
沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为四棱锥的密度;犮为四棱锥的底面边长;d为四棱锥的高度。
四棱锥密度为
式中:ε为四棱锥密度;S为每个四棱锥所占据的面积;犮为正四棱锥底面边长。
将式(1-38)代入式(1-37)得
式中 膜的比表面积;ε为四棱锥的密度;犮为四棱锥的底面边长;d为四棱锥的高度。
膜的比表面积;ε为四棱锥的密度;犮为四棱锥的底面边长;d为四棱锥的高度。
图1-21表示四方密排时正四棱锥高度和底面边长与比表面积之间的关系。
图1-21 四方密排时正四棱锥半径和高度与比表面积之间的关系
e.尖劈模型(e)是由正三棱锥以六方密排组成。设其底面边长为犮,高为d,则其比表面积为
式中:S膜为膜的比表面积;S表为尖劈膜的上表面积 为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为三棱锥的密度;犮为三棱锥的底面边长;d为三棱锥的高度。
为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为三棱锥的密度;犮为三棱锥的底面边长;d为三棱锥的高度。
三棱锥密度为
式中:ε为三棱锥密度;S为每个三棱锥所占据的面积;犮为正三棱锥底面边长。
将式(1-41)代入式(1-40)得
式中 膜的比表面积;ε为三棱锥的密度;犮为三棱锥的底面边长;d为三棱锥的高度。
膜的比表面积;ε为三棱锥的密度;犮为三棱锥的底面边长;d为三棱锥的高度。
图1-22表示六方密排时正三棱锥高度和底面边长与比表面积之间的关系。
图1-22 六方密排时正三棱锥半径和高度与比表面积之间的关系
根据前面对各种模型比表面积的计算可以看出除了平面模型外,其余模型的比表面积都随着其几何参数的变化而变化,而且都可以达到平面膜面积的几十到几百倍。为了便于分析,使得各种模型的参数d=R=犮,进行比较(见表1-3)。
表1-3 各种模型比表面积的比较
当按给定条件下d=R=犮,且纳米管的犽大于0.44时各种模型比表面积从大到小依次为:纳米管模型,尖劈模型(e),多孔模型,尖劈模型(d),尖劈模型(c),尖劈模型(b),尖劈模型(a),平面模型。当纳米管的犽值小于0.44时尖劈模型(e)表面积大于纳米管模型。
从前面对各种模型比表面积的计算结果可以看出,当多孔膜和纳米管模型的半径比较小,孔深比较大时其表面积可达原来沉积平面的几十到一百倍。当各种尖劈膜结构的底面半径比较小,高度比较大的时候其表面积也可以达到原来沉积平面的几十到一百倍。这几种膜结构在一定条件下都可以形成很大比表面积,但是在用于处理废水的光催化Ti O2膜有效的表面积是能与水接触的表面积,而且如多孔结构和纳米管结构的那些盲孔其随着孔的深度的增加,其表面积的有效性减弱。无论是空穴氧化理论还是羟基自由基氧化理论都要求催化剂与水接触,以达到空穴向污染物或者水转移进而氧化分解污染物。所以大的与水接触面积更有利于催化反应的进行。
膜与水的接触面积大小主要取决于膜与水的润湿性,而膜的润湿性由其化学组成和微观几何结构共同决定。本书中研究的是Ti O2膜,其化学组成已定。润湿性就主要由微观几何结构决定。通常以接触角θe表征液体对固体的浸润程度。在理想的固体表面上(结构、组成均一),接触角具有特定的值并由表面张力决定,满足Youngs方程,如图1-23所示。
图1-23 理想固体表面液体接触角与表面张力的关系
cosθe=(γsg-γsl)/γlg(1-43)
式中:γsg、γsl、γlg分别为固气、固液、气液间的界面张力。
真实固体表面在一定程度上或者粗糙不平,或者化学组成不均一。所以实际测定的表观接触角与Youngs方程预计值有较大的差异。Wenzel和Cassie等在20世纪40年代分别揭示了真实表面的非均一性对表面浸润性的影响,对Youngs方程进行了修正。当表面存在微细粗糙结构时,表面的表观接触角与本征接触角存在一定的差值,如表面的微细结构化可以将本征接触角为100°~120°的疏水表面呈现160°~175°甚至更高的表观接触角。Wenzel发现表面的粗糙结构可增强表面的浸润性,认为这是由于粗糙表面上的固液实际接触面积大于表观接触面积的缘故。于是在几何上增强了疏水性(或亲水性),他假设液体始终能填满粗糙表面上的凹槽,如图1-24所示,称之为湿接触,其表面自由能为
图1--24 Wenzel模型
d犌为三相线有dx移动时所需要的能量。平衡时可得表观接触角 和本征接触角
和本征接触角 之间的关系:
之间的关系:
cosθ=rcosθe(1-45)
式中:狉为实际的固/液界面接触面积与表观固/液界面接触面积之比其值大于等于1。
Wenzel方程揭示了粗糙表面的实际接触角 与
与 方程中的本征接触角θe之间有如下的关系:若θe<90°,则
方程中的本征接触角θe之间有如下的关系:若θe<90°,则 <θ,即表面的亲水性随表面粗糙程度的增加而增强;若θe>90°,则
<θ,即表面的亲水性随表面粗糙程度的增加而增强;若θe>90°,则 >θ,即表面的疏水性随表面粗糙程度的增加而增强。但因为
>θ,即表面的疏水性随表面粗糙程度的增加而增强。但因为 也只能处于0到180°之间,故θe>arccos(-1/狉)或θe<arccos(1-/狉)时
也只能处于0到180°之间,故θe>arccos(-1/狉)或θe<arccos(1-/狉)时 分别为180°和0°。Satoshi等通过实验所得到的数据,可以看出中间大部分较好地符合Wenzel的线性关系,也即印证了表面粗糙度也是调控表观接触角的主要因素,从此对表面结构化改变表面润湿性能有了较好的解释。但是在亲水区和超疏水区部分直线的截距并非为1或-1,而在到达1或-1附近有一个转折,其余数据点也近似为一直线,但其斜率显然变小很多。
分别为180°和0°。Satoshi等通过实验所得到的数据,可以看出中间大部分较好地符合Wenzel的线性关系,也即印证了表面粗糙度也是调控表观接触角的主要因素,从此对表面结构化改变表面润湿性能有了较好的解释。但是在亲水区和超疏水区部分直线的截距并非为1或-1,而在到达1或-1附近有一个转折,其余数据点也近似为一直线,但其斜率显然变小很多。
Cassie等研究了组成的不均一性对表面浸润性的影响,提出了复合接触的概念。即他们认为液滴在粗糙表面上的接触是一种复合接触。微细结构化了的表面因为结构尺度小于表面液滴的尺度,当表面结构疏水性较强时, Cassie认为在疏水表面上的液滴并不能填满粗糙表面上的凹槽,在液珠下将有截留的空气存在,于是表观上的液固接触面其实由固体和气体共同组成,如图1- 25所示。认为这种组成非均一表面的浸润性是各个组分浸润性的加和,表观接触角 )与各组分本征接触角(θ犻)的关系如下:
)与各组分本征接触角(θ犻)的关系如下:
图1--25 Cassie模型
式中: 犻是构成表面各组分的面积分数,
犻是构成表面各组分的面积分数, +
+ =1。
=1。
当表现为超疏水时,其中一种介质为空气时,其液气接触角为180°,得
式中: s为复合接触面中固体的面积分数,该值小于1,在疏水区该值越小表观接触角越大,该方程也可以通过表观接触角
s为复合接触面中固体的面积分数,该值小于1,在疏水区该值越小表观接触角越大,该方程也可以通过表观接触角 和本征接触角
和本征接触角 之间的关系表示。此线能较好地解释前面提及的接近超疏水区不符合Wenzel关系的那段直线,因此也可以看出,高疏水区域由于结构表面的疏水性导致液滴不易侵入表面结构而截留空气产生气膜,使得液珠仿佛是“坐”在粗糙表面之上,当表面足够疏水或者狉足够大时
之间的关系表示。此线能较好地解释前面提及的接近超疏水区不符合Wenzel关系的那段直线,因此也可以看出,高疏水区域由于结构表面的疏水性导致液滴不易侵入表面结构而截留空气产生气膜,使得液珠仿佛是“坐”在粗糙表面之上,当表面足够疏水或者狉足够大时 → 0
→ 0 → 180°,液滴将“坐”在“针”尖上。因此有效的计算参数只是固液接触面上固体表面所占的分数而不是粗糙度。
→ 180°,液滴将“坐”在“针”尖上。因此有效的计算参数只是固液接触面上固体表面所占的分数而不是粗糙度。
当表现为高亲水时,对于高亲水部分不符合Wenzel线性关系的直线也可以采用Cassie的复合接触理论来解释,微细结构化了的表面可以看作是一种多孔的材料,虽然这只是两维上的多孔但也显示出了与平坦表面不同的性能:当表面具有这种微细结构且具有较好的亲水性能时,表面结构易产生毛细作用而使液体易渗入并堆积于表面结构之中,所以此种结构易产生吸液而在表面产生一层液膜,但是并不会将粗糙结构完全淹没,仍有部分固体露于表面,所以再有液滴置于其上就会产生由液体和固体组成的复合接触面,其中一种介质为液体,相同液体间接触角为0°,得
式中 为表观接触面上固体所占的比例,很显然
为表观接触面上固体所占的比例,很显然 越小时表观接触角就会越小。液滴的三相线受到表面固体和结构中液体的共同作用而并非真正的圆形,这也是处于铺展(spreading)与吸液(imbibition)之间的一种状态如图1-26所示。按照Steven Garoff的建议,可以将之称为半毛细(hemi-wicking)作用。
越小时表观接触角就会越小。液滴的三相线受到表面固体和结构中液体的共同作用而并非真正的圆形,这也是处于铺展(spreading)与吸液(imbibition)之间的一种状态如图1-26所示。按照Steven Garoff的建议,可以将之称为半毛细(hemi-wicking)作用。
图1-26 犆b狊狊犻犲模型亲水表面液滴
本书中考虑的是各种不同结构Ti O2膜在浸入水中,具有一定静水压力的情况下其与水的接触面积。Ti O2与水的本证接触角约为72°,但当有紫外光照射时就会显示出高亲水性,即与水的接触角降到0°。A.Dupuis和J.M. Yeomans考虑表面能自由能,并利用格子Boltzmann算法求解Navier-Stokes方程。模拟在重力条件下半径为30的球形水滴在由长方柱(长4、宽4、高5)组成间距8的四方阵列上由悬浮态到浸入到阵列空隙的过程。长方柱与水的本征接触角为110°。张继华等将具有超疏水性的荷叶浸入50cm深的水中浸渍2小时,发现荷叶表面变湿。这表明即使是疏水材料组成的表面微结构,在有一定的静水压力时水也可以浸入到表面微结构的空隙之中。以上两个例子中的空隙之间相通,在这种情况下可能出现由于各点压强有所不同,压强大处的水进入空隙中将其中的空气挤压而排除,最终水进入到表面结构的微孔之中。但是对于如图1-27所示的微盲孔结构,由于孔径比较小,很容易被水将孔口封住。这时盲孔中的空气不能排除,水在静水压的作用下被压入孔中,孔中的气体由于被压缩气压增大。图1-27中R为水面曲率半径,狉为孔半径,(a)、(b)表示孔壁为疏水介质时的水面情况;(c)、(d)表示孔壁为亲水介质时水面情况。当孔壁为疏水介质时,即接触角大于90°,孔中的水面为凸球冠形,当接触角等于180°的极限情况时,孔中水面为以孔半径为半径的凸半球面。而当孔壁为亲水介质时,即接触角小于90°,孔中的水面为凹球冠形,当接触角等于0°的极限情况时,孔中水面为以孔半径为半径的凹半球面。所以对于微盲孔,无论孔壁为疏水还是亲水,水都只能在静压力的作用下部分进入孔中,而不能取代里面的气体完全填充孔。而且考虑到进入盲孔中的溶液与主体溶液之间的传质只能依靠扩散进行,这样随着孔深度的增加其盲孔内表面积的有效性则降低。
图1--27 微盲孔结构中水在一定静水压力存在下进入孔中
(a)接触角等于180°时,微盲孔中水面形成等于微孔半径的凸半球 (b)接触角大于90°小于180°时,微盲孔中水面形成大于微孔半径的凸球冠 (c)接触角大于0°小于90°时,微盲孔中水面形成大于微孔半径的凹球冠 (d)接触角等于0°时,微盲孔中水面形成等于微孔半径的凹半球
所以在计算有效比表面积时,对于盲孔结构只计算距开口处深度等于孔直径部分的孔内表面积,更深处的孔内表面积可以忽略。而对于空隙之间相互连通溶液可以流动的模型,则可以认为其表面积全为有效表面积。
①平面膜的有效比表面积。
根据前面的分析和对平面膜表面积的计算,对于平面膜,其有效表面积为1。
②多孔膜的有效比表面积。
根据前面的分析,多孔结构属于盲孔结构,故其孔壁的有效表面积只计算距开口处孔深度等于孔直径部分。如图1- 27所示,为便于计算假设多孔膜的孔为均一的,孔半径为R,密度为ε,则多孔膜的有效比表面积为
式中:S有效膜为膜的有效比表面积;S有效表为多孔膜的有效表面积;S沉积为沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为多孔膜的孔密度;R为多孔膜的孔半径。
从式(1-49)可以看出,多孔膜的有效比表面积主要与多孔膜的孔密度、孔半径有关。孔密度越大,孔半径越大,其有效比表面积也就越大。
将以密排六方排列时的最大孔密度式(1-24)代入式(1-49)得
这样对于多孔膜其有效比表面积为1 。
。
③纳米管膜的有效比表面积。
根据前面的分析,纳米管结构属于盲孔结构,故其孔壁的有效表面积只计算距开口处孔深度等于孔直径部分。如图1- 28所示,为便于计算假设纳米管是均一的,管外径为R,内径为狉,深度为d,纳米管膜的有效比表面积为(1-51)式中 为膜的有效比表面积
为膜的有效比表面积 纳米管膜的有效表面积
纳米管膜的有效表面积 沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为纳米管的密度;R为纳米管的外半径;狉为纳米管的内半径。
沉积薄膜所占面积;b为Ti O2膜所覆盖面积的长;犫为Ti O2膜所覆盖面积的宽;ε为纳米管的密度;R为纳米管的外半径;狉为纳米管的内半径。
图1--28 纳米管内外径比例与有效比表面积之间的关系
从式(1-51)可以看出,纳米管膜的有效比表面积主要与纳米管的密度、外半径、内半径有关。比表面积随着纳米管的密度、外径、内径的增大而增大。
与多孔膜结构类似,当纳米管以密排六方形式排列时其密度最大。将式(1-24)代入式(1-51)得
设犽为纳米管内外径之比:
k=r/R (1-53)
则
图1--28表示纳米管内外径比例与有效比表面积之间的关系。
从图1-28可以看出纳米管膜的有效比表面积随着纳米管内外径比值的增大而增大。随着内外径比值从0.001到0.999变化,有效比表面积的变化区间为1.91~2.81。
④尖劈结构膜的有效比表面积。
根据前面的分析,各种尖劈结构属于空隙间相互连通的开放性结构,故其有效比表面积与前面计算的比表面积一致。
尖劈模型(a)是由圆锥单元按四方阵列组合而成。其有效比表面积为
式中:S有效膜为膜的有效比表面积;R为圆锥的半径;d为圆锥的高度。
尖劈模型(b)是由圆锥单元按密排六方阵列组合而成。其有效比表面积为
式中:S有效膜为膜的有效比表面积;R为圆锥的半径;d为圆锥的高度。
尖劈模型(c)是由正六棱锥单元按密排六方排列而成。其有效比表面积为
式中:S有效膜为膜的有效比表面积;犮为四棱锥的底面边长;d为四棱锥的高度。
尖劈模型(e)是由正三棱锥以六方密排组成。其有效比表面积为
式中:S有效膜为膜的有效比表面积;犮为三棱锥的底面边长;d为三棱锥的高度。
根据前面对各种模型有效比表面积的计算可以看出除了平面模型、多孔膜型、纳米管模型外,其余模型的有效比表面积都随着其几何参数的变化而变化。为了便于分析,使得各种模型的参数d=R=犮,进行比较(见表1-4)。
表1--4 各种模型有效比表面积的比较
不同形貌的Ti O2膜模型不但具有不同的表面积、有效比表面积,其对光的吸收率也不尽相同。采用光线追迹技术对Ti O2膜模型进行模拟,从而得出不同膜结构的Ti O2膜对光的吸收率。
在几何光学中,当光线到达两种光介质的界面时,入射光线就会被分为反射光线和折射光线。假定入射光线是从空气向一种光密介质传播,这种光密介质对光线有一定的吸收作用,当折射光在这种光密介质中传播时就会被其吸收。如果界面是平面时,反射线反射回空气中,不能被吸收。然而,当我们创造一种具有侧面的界面,如金字塔结构界面,如图1-29所示。如果一些几何关系满足时,当从一个侧面反射回来的反射光线会入射到另一个侧面。而且可以看出当金字塔结构顶角减小时,反射次数就会增加,每多一次反射就多一次吸收,这样就会增加光的吸收率。假如反射光线与折射光线强度相等,也就是入射光线强度的一半,反射次数为n,由于所有折射光线被光密介质完全吸收,最终反射进入空气的n次反射线的强度就为入射光线强度的0.5n倍。当n增加,最终反射光线的强度就会急剧下降。这就是光陷阱结构的陷光原理。
图1--29 二维光陷阱结构
(a)二次入射,θ=90° (b)三次入射,θ=60° (c)多次入射,θ=30°
模拟过程是在Lambda Research公司基于光纤追迹技术的Trance Pro软件中进行。模拟过程如图1-30所示。用于研究的具有光陷阱结构的尺寸为500μm×500μm,厚度为100μm。光源为300μm×300μm,光源波长从300~1200nm。波长进步长度为10nm,光线总数为10000条,总能量为1W。光从垂直于表面方向照射到模型表面。光线经光密介质表面的多次反射吸收,使最终反射出其表面的光线能量降到很低。反射率数据是用分光光度计测试不同波长的光在机械抛光硅表面的反射得出,光波长度范围为300~1200nm,如图1-31(a)所示。太阳光在不同波长的辐射强度数据如图1-31(b)所示。最终,结构吸收率定义为
图1--30 光线追迹过程
实线为入射光线;虚线和点画线为反射线。不同类型的线表示其强度不同。实线、虚线、点画线的强度依次减弱。
式中 为结构在一定顶角和一定波长时的吸光率
为结构在一定顶角和一定波长时的吸光率 为最初入射光的总强度;犐1为最终反射出光密介质表面的光强度。不同光陷阱结构在不同顶角对太阳光的地吸收率用下式计算:
为最初入射光的总强度;犐1为最终反射出光密介质表面的光强度。不同光陷阱结构在不同顶角对太阳光的地吸收率用下式计算:
式中 为一定角度时对太阳光的吸收率;犃犻为波长为犻nm时的吸收率;S犻为太阳光在波长为犻nm处的辐射强度。
为一定角度时对太阳光的吸收率;犃犻为波长为犻nm时的吸收率;S犻为太阳光在波长为犻nm处的辐射强度。
为了评估光线追迹模拟技术的可靠性,对比了模拟值与实验实测值。模拟值是使用光线追迹技术模拟与化学腐蚀形貌类似的顶角为70°的四棱锥结构得出;实验值是使用分光光度计实测出,由Na OH溶液对单晶硅进行了各向异性腐蚀,制备出的具有四棱锥结构的绒面硅的反射率。结果如图1-31(a)所示,对比模拟值和实验值可以看出,反射率的模拟值与实验测试值非常接近。模拟曲线趋势与实验曲线完全符合。模拟值比实验值稍低一些。这是因为用于模拟的结构与化学腐蚀硅的结构不完全一样。用于模拟的结构是非常理想和规整的,而化学腐蚀结构不规整。所以可以认为本书中用到的光线追迹模拟技术是可靠的。
图1-31 (b)抛光硅、化学腐蚀硅表面对不同波长光的反射率
测试值和化学腐蚀硅表面对不同波长光的反射率模拟值 (犫)到达地面的太阳辐射光谱
①平面膜、多孔膜和纳米管膜的吸光率。
平面膜、多孔膜和纳米管膜模型的吸光率模拟结果如图1-32所示。这几种模型的吸光率与抛光面的吸光率相等。由于这几种模型都没有倾斜的表面,所以当光线垂直于其表面入射时反射光线将不能再次入射其表面形成吸收,所有光线只有一次吸收。
图1-32 平面膜、多孔膜和纳米管膜模型对不同波长光的吸光率
②尖劈结构膜的吸光率。
光陷阱结构密度对其吸光率的影响如图1-33所示。吸光率用等高线表示。四方阵列圆锥结构和密排圆锥结构的圆锥覆盖率分别为89.1%和96.1%。从图1-33中可以看出,图1-33(b)中吸光率在0.8~0.9的面积大于图1-33(a)。而且在图1-33(b)中有一个小面积代表吸光率达到0.9~0.9999,而图1-33(a)中没有出现代表吸光率达到0.9~0.9999的面积。这表明高的圆锥覆盖率具有高的光吸光率。因此,吸光率随着圆锥覆盖率的增大而增大。这就意味着表面光陷阱结构密度越大其吸光率越高,这与光陷阱结构的陷光原理一致。
图1--33  四方阵列
四方阵列  六方阵列排布的圆锥结构模型在不同顶角和不同波长时的吸光率
六方阵列排布的圆锥结构模型在不同顶角和不同波长时的吸光率
光陷阱结构的几何形状对吸光性能的影响如图1-33、图1-34所示。当光陷阱结构单元从圆锥、六棱锥、四棱锥、三棱锥依次变化时,图中表示吸光率为0.9~0.9999的面积依次增大。当顶角小于100°三棱锥光陷阱结构对波长在640~1080nm的光的吸收率都超过90%。这表明在这些光陷阱结构中,三棱锥光陷阱结构表现出最好的吸光性能,而圆锥光陷阱结构的吸光性能最差。当光陷阱结构从三棱锥过渡到圆锥,吸光率减小了。这是因为三棱锥的斜面比其他结构能捕获更多的反射光线。
图1--34 不同结构模型在顶角和不同波长时的吸光率
(a)六棱锥 (b)四棱锥 (c)三棱锥
顶角对光陷阱结构的吸光率的影响如图1-33、图1-34所示。当圆锥、六棱锥、四棱锥和三棱锥的顶角分别大于110°、115°、130°和145°时,其吸光率与机械抛光硅一样。这是因为在这些情况下,光线到达其表面一次以后就反射出表面。斜面的倾斜度太小而不能在其表面产生多次反射,这样就不能构成光陷阱结构。当圆锥、六棱锥、四棱锥和三棱锥的顶角分别小于70°、85°、100°和145°时,其对波长在640~1080nm光的吸收率均大于0.9。这是因为随着顶角的减小光在光陷阱结构表面的发射次数增加如图1-29所示。在这种情况下,光在光陷阱结构表面反复反射,最终几乎全部被吸收。
不同光陷阱结构在不同顶角对太阳光的吸收率计算结果如图1- 35所示。对太阳光的吸收率大小依次为三棱锥、四棱锥、六棱锥、密排圆锥和正方阵列圆锥光陷阱结构。当顶角分别小于60°、80°、100°六棱锥、四棱锥和三棱锥光陷阱结构对太阳光吸收率相同,均大于85%。三棱锥光陷阱结构的范围要比其他两种光陷阱结构宽。所以在这些光陷阱结构中三棱锥光陷阱结构具有最好的光陷阱效应。而正方阵列圆锥光陷阱结构的光陷阱效应最差。甚至当顶角已经足够尖,小于60°,四方阵列圆锥光陷阱结构对太阳光的吸收也不高,小于80%。硅对光的吸收率与硅表面光陷阱结构单元的形状、顶角和密度3个因素相关。其中密度是影响最大,其次是角度和形状。这与前面对密度、角度和形状对吸收率的影响的讨论结果一致。
图1-35 不同结构模型在不同顶角时对太阳光的吸光率
(a)三棱锥 (b)四棱锥 (c)六棱锥 (d)六方密排圆锥 (e)四方排列圆锥
根据对各种模型吸光率模拟结果可以看出各种模型吸光性能由好到差依次为:尖劈模型(e),尖劈模型(d),尖劈模型(c),尖劈模型(b),尖劈模型(a),纳米管模型、多孔膜性、平面模型。
通过计算和模拟,可以得出三棱锥结构的微结构模型既具有较大的有效比表面积,同时具有高的光吸收率,从而具有较高的光催化效率。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










