



三、常用算法
(一)分子力学方法
分子力学方法(molecular mechanics,MM)是一个广泛应用于计算分子几何构型和能量的方法。它将原子看作大小不同的橡皮球,把键看作长度不同的弹簧,不考虑电子和核,将蛋白质分子简化成作简谐振动的力学模型,利用Hook定律计算原子势能。其优点是计算速度非常快,适用范围广。其能量函数通常用一系列能量项的和来表示:
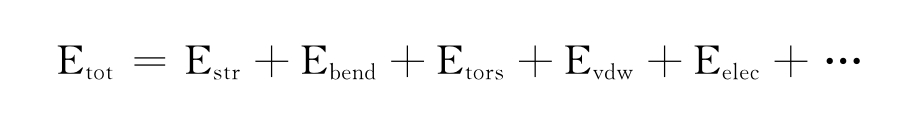
其中:Etot代表分子总能量;Estr代表键伸缩能量项;Ebend表示键角扭转能量项;Etors表示二面角扭转能量项;Evdw表示范德华能量项;Eelec表示静电作用能量项。
在蛋白质三维结构模拟中,MM算法常用的有最陡下降法(steepest descent)和共轭梯度法(conjugate gradient algorithm)。
(二)分子动力学方法
分子动力学(molecular dynamics,MD)以分子力学为基础,在给定的势能函数和力场下对经典牛顿力学运动方程积分,从而搜索蛋白质分子系统的运动和构型空间。其基本假设是:对于无限空间的平均等于对系统整体的平均或者对构型空间的积分。
Fi(t)= miai(t)
Fi(t)=-▽iE
miai(t)= miδ2xi(t)/δt2
常用的积分算法有Verlet积分、Beeman算法和Leap-Frog算法。分子动力学最初用于模拟简单液体行为,计算热力学性质,后来扩展到模拟多肽、晶体状态和溶液中的蛋白质,以及低聚糖、寡糖等。
分子动力学用于构象搜索时,用势能函数的梯度▽iE来表示力Fi,随机产生一个初速度,以原子的初始坐标为起点,计算原子在t时刻的新位置和速度,产生新构象,在给定的时间内多次迭代,便可以获得分子的优势构象。产生新构象的时间间隔通常取1~2ps(picosecond,10-12s)。
相比于分子力学方法,分子动力学方法更能够克服不同构象之间的能垒,从而达到更广的构象搜索空间。
(三)量子力学方法
蛋白质分子结构中有些特殊的结构往往没有合适的力场参数可供选用,此时,可用量子力学方法(quantum mechanical,QM)优化。而且当涉及到反应过渡态、极化、特殊电子云分布、分子轨道改变时,只能采用量子力学方法计算。其不足之处是计算代价太大,需要耗费大量的计算机内存和计算时间。
量子力学方法是基于分子轨道理论(molecular orbital theory),并以下列3个近似为出发点的:①采用非相对论的量子理论,即基于薛定谔(Schr9dinger)方程。②采用玻尔奥本海默(Born-Oppenheimer)近似,即将核运动和电子运动分离开来。③采用轨道近似,即以原子轨道的组合表示分子轨道。
(四)构象分析和搜索
蛋白质分子构象的几何优化可以使分子获得相应于初始态的优势构象,但蛋白质分子往往存在许多种优势构象,分子结构在不同的优势构象之间变换,从而处于一种相对的平衡状态。从一个构象到另一个构象的转变主要是由于二面角的变化,而键长、键角的改变很小。分子构象的改变可以看作是沿势能表面的运动,每一点代表相应的势能及其构象,势能极小点即为稳定构象。一般情况下,普遍认为蛋白质分子拥有多个稳定构象,亦称为蛋白质分子的柔性特征。
当外界环境变化,例如发生酶与底物的相互接近时,蛋白质分子的构象会发生较大的变化,变化的结果是形成酶底物复合物或过渡态、中间态。蛋白质分子与药物或其他配体作用时的情况与此相类似,普遍认为药物分子与受体蛋白作用时,总是采取某一低能构象,称为活性构象或药效构象。一般情况下,活性构象并不是能量最低构象,但也不可能是能量最高构象,因此,低能构象的识别可确定药物与受体作用方式,并以此作为基础,设计新药。
对蛋白质分子的稳定构象和低能活性构象的识别需要借助理论计算方法。最直接的方法是识别势能面的所有极小点,但是,由于蛋白质分子属生物大分子,分子里可旋转键的数目庞大,造成势能面上的极小点急剧增加,穷尽搜索几乎是不可能的。为此,发展了许多构象搜索方法。
由于量子力学计算非常耗时,不能用于生物大分子和高柔性分子,因此,大多数构象搜索方法以分子力场计算能量。分子动力学计算,其本质上就是一种构象搜索的方法。
构象搜索的方法目前主要有系统搜索法、Monte Carlo法、模拟退火算法(SA)、遗传算法(GA/GAs)以及人工神经元网络算法(NN)。
1.Monte Carlo法(MC) 蛋白质模拟中常用Monte Carlo法。它不同于确定性数值方法,用于解决数学和物理问题的非确定性的(概率统计的或随机的)数值方法,故也称统计试验方法。它是理论物理学两大主要学科的合并:即随机过程的概率统计理论(用于处理布朗运动或随机游动实验)和位势理论,主要是研究均匀介质的稳定状态,用一系列随机数近似解决问题。通过寻找一个概率统计的相似体,从实验取样过程获得该相似体的近似解,处理数学问题。运用近似方法获得的解更接近于物理实验结果,而不是经典数值计算结果。
当前应用的MC技术,早在1944年已开始研究。MC技术的发展归功于早期核武器的研究,Los Alamos(洛斯阿拉莫斯美国国家实验室中子散射研究中心)的基础工作刺激了MC技术的形成和发展,并使MC应用于各种问题中的研究。
Monte Carlo方法的应用有2种途径:仿真和取样。仿真是指提供实际随机现象的数学上的模仿的方法。一个典型的例子就是对中子进入反应堆屏障的运动进行仿真,用随机游动来模仿中子的锯齿形路径。取样是研究少量的随机的子集演绎大量元素的特性的方法。例如,f(x)在a<x<b上的平均值可以通过间歇性随机选取的有限个数点的平均值进行估计。即数值积分的Monte Carlo方法。MC已成功地用于求解微分方程和积分方程,求解本征值,矩阵转置,以及计算多重积分。
任何本质上随机组员的过程或系统的仿真都需要一种产生或获得随机数的方法。这种仿真的例子在蛋白质折叠、配受体分子对接、中子随机碰撞、数值统计、队列模型、战略游戏、博彩以及其他竞赛活动中都会出现。Monte Carlo计算方法需要有可得的、服从特定概率分布的、随机选取的数值序列。
2.模拟退火算法 核心思想与热力学的原理颇为相似,本质在于对物质缓慢地制冷以争取充足的时间,让大量原子在丧失可动性之前进行重新分布。
3.遗传算法 借鉴生物界自然选择和进化机制发展起来的高度并行、随机、自适应搜索算法,特别适合于传统搜索算法解决不了的复杂的和非线性问题。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










