



第三节 致密沉积物病
致密沉积物病(dense deposit disease,DDD)又称Ⅱ型膜性增生性肾小球肾炎(membrano-poliferative glomerulonephritis typeⅡ)或Ⅱ型系膜毛细血管性肾小球肾炎(mesangio-capillary glomerulonephritis typeⅡ),其病理特征主要是电镜下发现GBM致密层内有均质电子致密物(electron-dense material)沉积,故也有膜内致密沉积物病(dense intramembranous deposit disease)之称。
本病于1963年,首先由Berger和Galle报道,此后在欧洲、美国及我国台湾省等地有相继报道,我组张志刚等于1999年在国内首先报道1例,2001年王梅等又报道5例。本病在我国较为少见,我组至今仅发现3例。
本病多发生于青少年或壮年,约70%的病例以肾病综合征为表现,少数则可表现为急性肾炎综合征或发作性血尿,90%以上的病例可伴镜下血尿,约65%病例可出现血浆持续性补体C3水平下降,也有约60%的病例可检出血浆内存在C3致肾炎因子(C3 nephritic factor,C3NeF)。
【病因和发病机制】
DDD的病因和发病机制均不明。据文献报道,该病可伴发于某些细菌(如链球菌、肺炎球菌等)感染,故提示其可能为一种少见类型的免疫复合物病,但其在肾小球等部位沉积的电子致密物仅含补体C3,而从未发现有任何免疫球蛋白成分,因此对其性质尚未作出肯定结论。
据最初一些学者对本病的研究,认为其与GBM原发性代谢异常有关,因为经对沉积致密物的分析,发现其含有不饱和脂肪酸复合物,又经进一步生化分析,发现其比正常GBM富含涎酸而胱氨酸较少,且证明其含有N-乙酰葡萄糖胺(N-acetyl glucosamine)成分。但以后的一些学者发现,这种致密物沉积现象也可在移植肾中发现,从而提示本病的发生可能与某些循环因子有关。而另一些作者则根据本病多数患者血浆中,出现一种属于IgG类的自身抗体,即C3NeF,可结合并稳定血浆内的C3转换酶(C3-converting enzyme)或称C3裂解酶(C3-splitting enzyme)(即为C3bBb),可经旁路持续地激活C3,生成产物C3b,进而生成更多的C3bBb,导致部分患者血浆C3水平呈持续地下降,而C1和C4的水平却保持在正常范围,故认为其发病与C3NeF有关。但因部分患者的血浆C3水平属于正常,而且在对家兔的实验中,也不能证实采用持续性旁路激活C3的办法,复制成DDD动物模型,因此,对C3NeF在DDD发病中的作用,目前也难以作出结论。至于在某些患者的肾组织内,出现肾小球上皮下“驼峰状”电子致密物沉积的改变,一些作者则认为,是因重叠了免疫复合物沉积所致。
【病理改变】
病变早期,约10%的病例因光镜下仅显示肾小球局灶节段性系膜增生或毛细血管壁轻度增厚的改变(图5-26)而易被漏诊。但在绝大多数病例中,肾小球在光镜下表现为膜性增生性肾炎或非典型性膜性肾炎(图5-27),肾小球血管襻呈嗜伊红性,缎带样(ribbon-like),经PAS和PASM染色显示GBM明显增厚(图5-28),或呈现节段性双层结构(图5-29)。在约2/3病例中,可伴有系膜细胞增生和弥漫性系膜基质增多,以轻-中度为多见,偶然呈重度,乃至形成系膜区结节。约2/3患者的肾小球内有新月体形成,部分病例的肾小球血管壁可发生纤维蛋白样坏死、中性粒细胞浸润和肾小球硬化等病变。肾小管可呈灶性萎缩,伴有肾间质炎症细胞浸润及纤维组织增生。
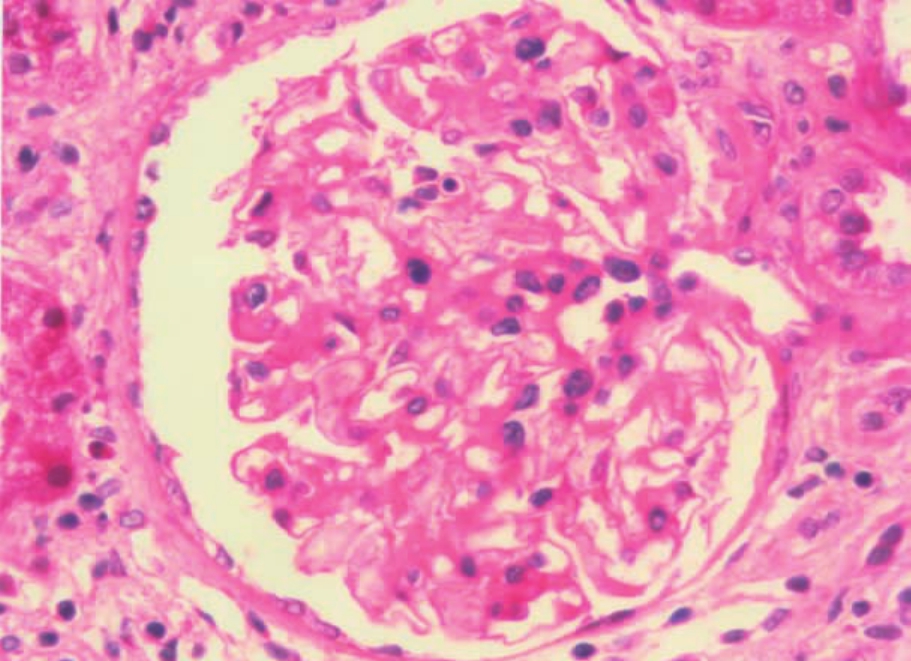
▲图5-26 致密沉积物病(HE ×400)
肾小球部分毛细血管壁增厚,伴轻度系膜基质增多

▲图5-27 致密沉积物病(HE ×200)
肾小球毛细血管壁增厚,伴系膜细胞中度增生及基质增多

▲图5-28 致密沉积物病(PAS ×200)
与图5-26为同一病例,肾小球部分毛细血管基膜增厚,伴部分系膜区基质增多

▲图5-29 致密沉积物病(PASM ×400)
与图5-27为同一病例,毛细血管系膜基质增多,基膜增厚,伴部分双轨状改变
免疫荧光检查可显示C3呈持续或断线状沿GBM沉积,弱的粗细颗粒状荧光也可出现于肾小球系膜区。C3呈阳性的免疫荧光也可出现在肾小球囊壁和近端小管基膜。可伴有IgM沉积,但IgG、IgA的沉积则十分少见。据统计,约1/3患者未发现任何补体和免疫球蛋白在肾小球内的沉积。
电镜检查是确诊DDD必不可少的手段,其典型而特征性改变是GBM致密层内有“缎带”状或融合性团块状、均质高电子密度的致密物沉积(图5-30,5-31),偶然可随血管襻轴心部的GBM皱折而出现在系膜区。GBM常因此而增厚,其厚度可达1 500 nm。
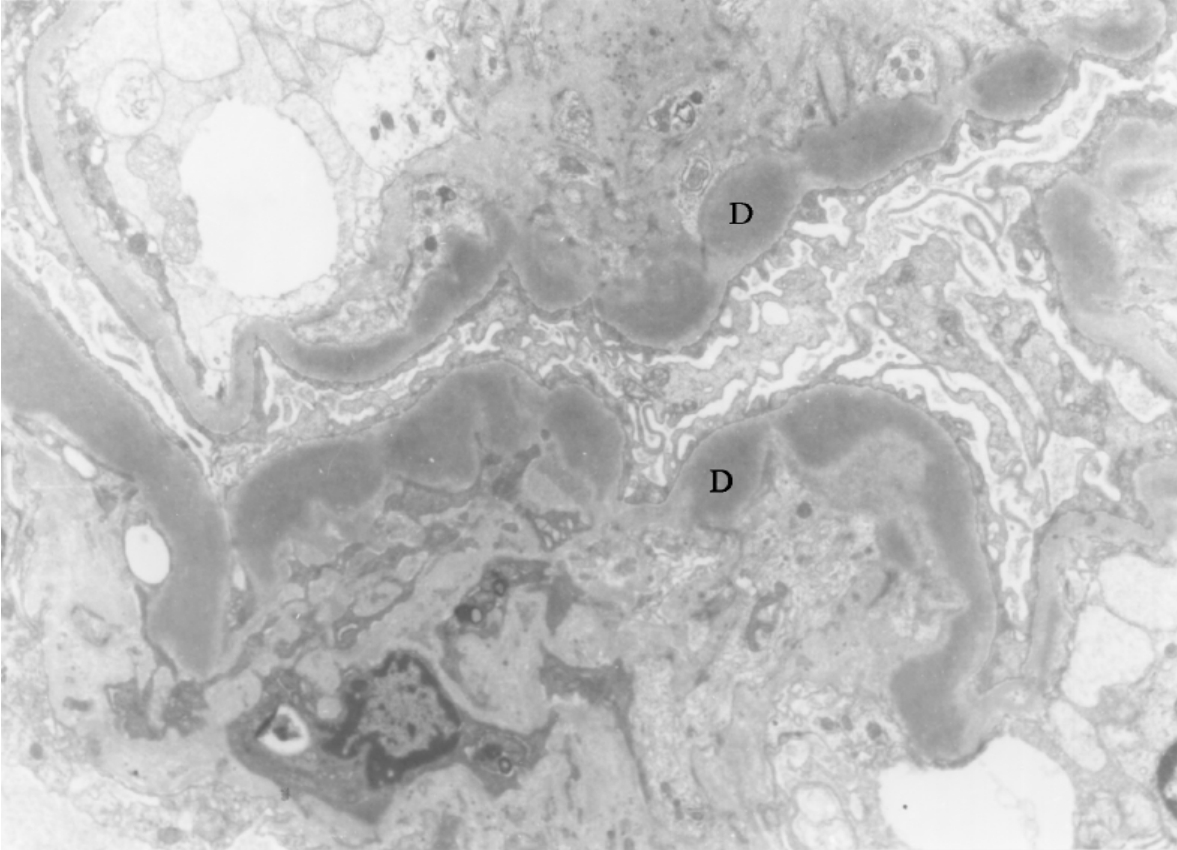
▲图5-30 致密沉积物病(EM ×7 500)
与图5-26为同一病例,肾小球血管壁基膜致密层内有呈连续、带状电子致密物(D)沉积
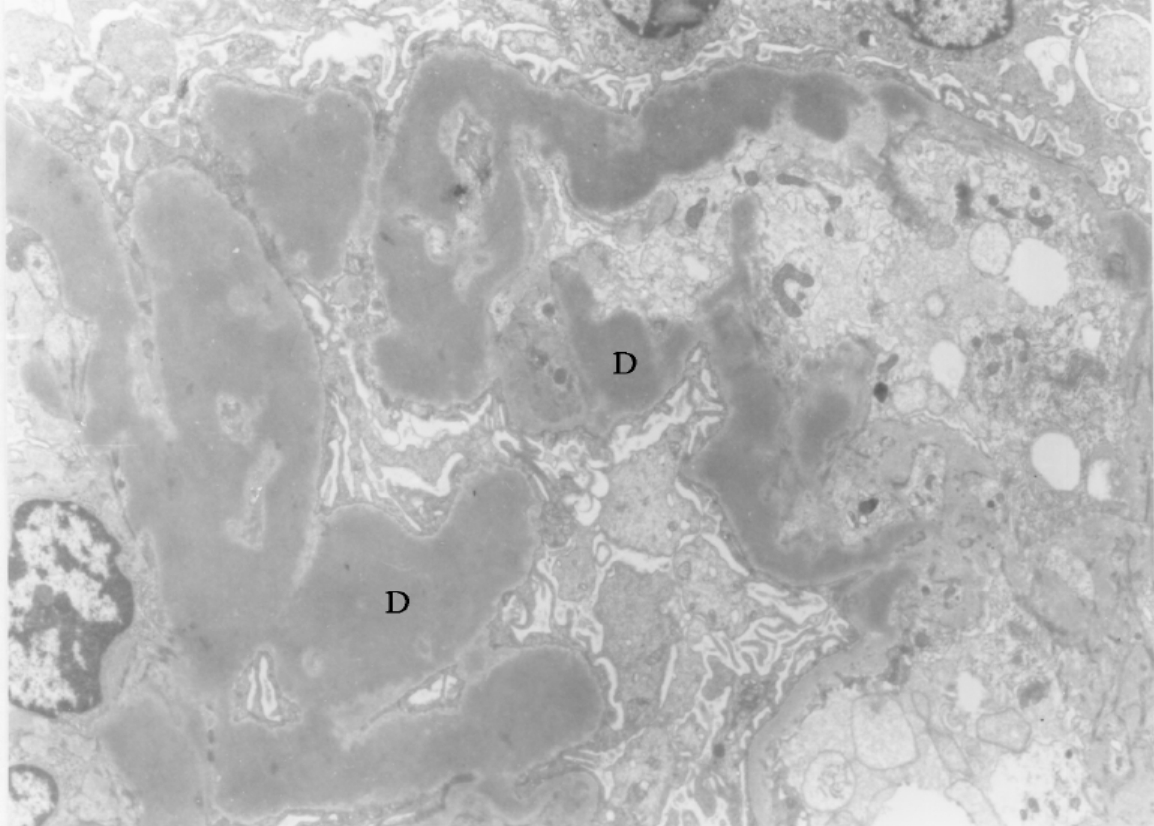
▲图5-31 致密沉积物病(EM ×6 000)
与图5-27 为同一病例,肾小球血管壁基膜增厚,致密层内呈现不均匀、带状电子致密物(D)沉积
【鉴别诊断】
DDD因其在电镜下显示,GBM致密层呈现特征性高电子密度致密物的沉积,其诊断并不困难。须与其作鉴别者首先是轻链肾病,但后者的电子致密物主要沉积于GBM的内疏松层,且免疫病理检查显示肾小球内有免疫球蛋白轻链(κ或λ)的沉积。与其鉴别的另一种肾小球病,则是伴有膜内沉积的Ⅲ型膜性增生性肾炎,但其沉积物不呈高电子密度性、GBM常有断裂,免疫荧光检查显示肾小球内有免疫球蛋白和其他补体成分的沉积。
【预后】
DDD是一种呈慢性进行性的肾小球病。据文献报道,患者经10年后,则进入慢性肾衰竭而需行血液透析治疗。若在病程的任何阶段出现急性肾损害,这就意味着其病理类型发生转变,最常见者为新月体性肾炎。对DDD实施同种异体肾移植者,其复发率甚高,据一作者对48例患者资料的统计,其中42例出现该病的复发。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










