



第三节 高聚物的性能
由于高分子化合物的巨大分子量,加上链间作用力又很大,使高分子链的运动出现很多低分子不具有的特性,从而使高聚物出现一系列独特的热运动特征及力学特征。
高分子链具有侧基、链节、支链、链段等不同层次的结构单元,因而存在与这些结构单元相应的各种运动,在一定温度下还可出现数根大分子链的协同运动,这就使高分子链的运动带有多样性。链段运动和整个大分子链的运动在此起着主要作用,因而习惯上称高分子的运动具有两重性。链段运动主要表现在高聚物的弹性变形过程中,整个高分子链的运动则主要出现于流动变形的不可逆过程中。在外力作用下,链段由原来的构象过渡到与外力相适应的构象时,需要时间来克服链段间的内摩擦所造成的粘性阻力,因而高分子的运动对时间有很大的依赖性。这就使同一种高聚物在同一温度下,使外力作用速度的不同,表现出不同的力学状态。温度对力学状态的影响比作用力的速度更为显著。温度升高使单键的内旋转加剧,首先是局部原子基团和小链段的短程振动增强,随后出现链段的长程运动,逐步扩大到相邻高分子链的相对滑移,进入粘性流动状态。这一过程中高聚物经历了力学状态的系列变化,总体表现为体积松驰过程。因此高聚物的聚集态的转变不是典型的热力学相变。
二、高聚物的流变学性质
流变学是研究物质形变和流动的科学。聚合物的流变行为包括几种差异很大的现象,这些现象构成了描述线型非晶态高聚物的力学性能的基础。部分结晶的高聚物的力学性能与结晶部分的性质密切相关,因而只能部分地从非晶态高聚物的流变学行为加以推测。
1.高弹性
高弹性是橡胶一类的主链柔顺的非晶态线型高聚物特征的物理状态(部分晶态聚合物也有此特征)。在此状态下。链段能在外力作用下发生长程运动(链段结构的存在及链段自由运动方式是高聚物特有的),使链段构象过渡到与外力相适应的状态,但是由链缠结或交联构成的散乱网状结构又会阻碍整个大分子链的运动。链段间发生相对运动时的内摩擦使弹性形变需要一定的时间才能完成。这种过程称为弹性松驰;该过程所需的时间称为弹性松驰时间,用t表示。高弹性是高聚物独有的特征。
高聚物弹性体的主要力学特征为:弹性形变量大,可达1000%;弹性松驰现象在充分拉伸时仍有较高的抗张强度及模量,外力去除后回弹至原有平衡状态(回缩性现象)。弹性优异的高聚物的弹性松驰时间短,即使在外力作用时间短、速度快的情况下也能显示出弹性性能。它在受力时易于发生形变,且形变量大,外力消除后能很快回复原有状态。(如图5.4)

图5.4 高聚物的力学状态与分子量、温度的关系示意图
弹性松驰现象在橡胶的弹性形变中影响较大。温度升高时,大分子链段的热运动加剧,外力作用下松驰过程较易进行,使松驰时间缩短。换言之,在相同外力作用时间下,温度升高使弹性形变增大。在温度与外力恒定时,如果外力作用速度极快,超过高分子链的松驰速度,则高聚物不呈现高弹态所具有的弹性形变,甚至可能发生大分子链的断裂。只有外力的作用时间接近或超过高聚物弹性体的弹性松驰时间,弹性才较好显示。升高温度和提高作用力速度对弹性形变具有相反的影响。
橡胶类的高聚物弹性体与金属相比,其弹性性能除了弹性形变大及弹性模量低(约为钢的1/106)等数量差异外,还有下列独特性:弹性模量随温度增大,这与钢钵的性质正好相反;与一般固体的内能弹性不同,高聚物的弹性来自柔顺性大分子链的构象改变,仅与熵值变化有关(称为熵弹性),即在外力作用下高分子链从处于未拉伸状态的最可几的构象形态出发,产生构象转变,但内能无变化,这种现象类似于理想气体的弹性特征;在伸长率不大时,绝热形变过程伴有明显的热效应,绝热位伸时放热,回弹时吸热。
2.粘弹性
将未硫化的天然橡胶在恒温下尽可能地缓慢拉伸,然后缓慢撤去外力,橡胶发生回缩,但回缩曲线的形变严重地落后于应力的降低,这种现象称为滞后。图5.5示经两个拉伸一回缩循环的形变应力曲线。图中,在第一循环开始时,用伸长率表示的形变以近似恒定的斜率随应力而迅速增加。当伸长率达100%左右时,橡胶在恒定应力下仍继续缓慢伸长,表现出塑性形变特征。降低应力时橡胶回缩,回缩曲线位于拉伸曲线下方。它与拉伸曲线共同构成一伸缩回线,称为滞后圈。
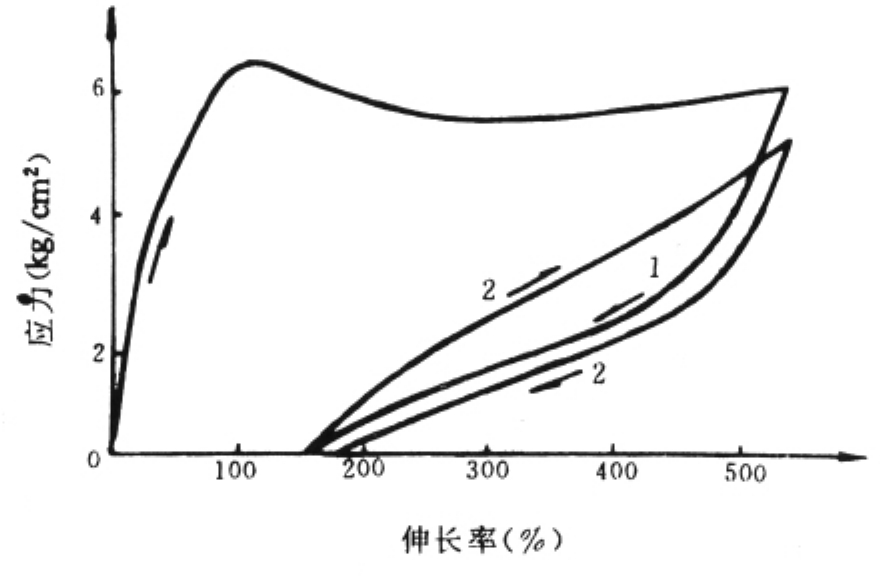
图5.5 天然橡胶的拉伸应力-形变曲线
实验表明,在首次拉伸时,橡胶己部分取向及结晶,因而取消应力后高分子链的结构不能充分回复原状,使橡胶在第二次伸缩循环时的弹性较为突出,较少塑性形变,滞后现象及永久形变也大为减小。与第二次伸缩循环相应的滞后圈的面积也变小。
继续增加伸缩循环次数,会使橡胶内部聚集态结构的变化固定下来,表现为滞后现象越来越弱。
通常的备向同性的理想弹性固体的形变,在形变很小时为瞬时的可逆形变,应力与应变呈线性关系,不依赖于应变速率,这类弹性称为平衡弹性。以形变较大时的天然橡胶为代表,高分子材料的固体形变特征与理想弹性固体的不同,在外力作用下,大分子链通过逐渐调整构象来抵消一部分外力,因而在弹性中伴有协同发生的粘流性,表现为兼具粘性的粘弹性体。撤去外力后,形变不能立即消除,有时甚至出现一定的永久形变。它的形变一应力关系呈显著的时间依赖性。固态高聚物的力学性能随时间而变化的现象总称为“力学松驰”或“粘弹性”。
应力不随时间而改变的力学松驰现象称为静态粘弹现象。它有蠕变及应力松驰两种表现形式。应力为时间的函数时,则称为动态粘弹现象。它有滞后和内耗等表现形式。
蠕变是材料的冷流现象,在固定外力下,材料的相对形变随时间而发展,经过一段时间后才能达到最大形变。蠕变通常是松驰和粘性流动的综合结果。前者涉及大分子的结构改变,所需要的时间长;后者表现为高聚物的形状改变,涉及时间短。对于小的应变,松驰现象占据主要地位,此时可以把应力松驰与蠕变定性地联系起来。随着形变总量的增大,粘性流动现象趋于显著。高分子材料的蠕变远超过金属及其它非金属材料,会破坏固体高聚物的尺寸稳定性。不利于高聚物的使用。蠕变性依赖于温度和负荷。在玻璃化转变温度以下,许多高聚物的蠕变性并不显著,即使经受很长时间的外力作用,也只发生极小蠕变。随温度升高,蠕变速度会增大。在玻璃化转变区域内,蠕变性质强烈依赖于温度,蠕变速度在玻璃化转变点附近出现一个极大值。温度继续升高,在玻璃化转变温度以上,伸长会大得多,但是蠕变速度通常会降低。负荷增大会使材料的蠕变倾向增大。蠕变性与高聚物的组成和结构有关。通常,主链含有芳环的高聚物及主链比较僵硬的高聚物的抗蠕变性较好。通过透变的交联及纤维增强,有利于减小高分子材料的蠕变性。结晶高聚物的蠕变行为更复杂,其蠕变性不仅随温度作较快变化,且在给定温度下,有时结晶高聚物随时间的蠕变反而强于非晶态硬聚合物及交联聚合物,这种现象可能来自结晶度随温度的变化、玻璃化转变温度与熔点的相对差距的大小以及可能的再结晶现象。
在一定形变下,高分子材料内部产生较大应力。通过缓慢调整大分子构象或位移,内应力逐渐衰减,大分子链趋于自然卷曲的稳定状态,在此过程中制品的形状并无改变,这是高分子材料的应力松驰的特点。衰减的应力消耗在两方面:松动大分子链间的部分缠结;克服大分子链的构象调整过程中的粘性内摩擦力,这种力阻碍了链段的自由运动。应力松驰现象可以视为热运动对大分子取向的影响。作用在高聚物上的机械应力引起链的形变。由于构象几率的减小会使体系的熵减小,自由能则相应增加。应力松驰的发生是链的热运动的结果。它不同程度地消除了大分子的变形,过剩的自由能则以热能形式耗散。应力松驰的细节过程有多种途径,总的表现为通过热运动使大分子恢复到最可几的构象。高聚物分子的这些复杂运动,可以用大分子链中链段的不同程度的系列长程协同运动的特征形式来描述,其中,对应于整个分子的移动的是最大的协同运动。在橡胶态区域中,链缠结是重要的,这个区域的分子运动基本上是长区域的运动,包括长度与大分子同数量级的单元的运动。因此,柔顺的大分子链易于发生应力松驰;经过足够长的时间后,未交联的天然橡胶的应力可衰减至零。经一定的交联后应力松驰被大大抑制,应力只能衰减至某平衡值,而不会降低到零在玻璃化转变区域附近,只有位于缠结点间的分子链的局部分子振动才是重要的,已经证实此时的应力松驰主要与聚合物局部结构有关,而与分子量无关。此区域内应力松驰强烈依赖于时间。当温度达到Tg或Tg以下时,模量的衰减明显地平缓下来。
橡胶类材料用于滚动轮胎、传送带、吸收震动的减震器时,要在正弦性交变应力下工作,产生伸一缩循环应变。由于大分子的链段运动是一种需要时间的过程,应变的发展必然落后于应力,使应变与应力间构成相位差。当使用频率或应变速率较高时,分子链来不及发生构象重排便出现内摩擦,从而导致额外的能量损耗(即内耗)。在理想的动态形变情况下,拉伸功与回缩功应相等。但是由于塑性永久形变的发展,部分拉伸功消耗于克服大分子链间的内摩擦和链段取向,变成热量向周围散失,使非理想的动态形变的回缩功必小于拉伸功,这两者间的差额称为“滞后损失”。滞后现象是否出现与交变应力的频率密切相关。交变应力频率很低时,大分子链有足够时间通过一系列的链段运动改变其构象,以适应交变外力,此时不存在滞后现象。交变应力频率很高时,链段运动远落后于应力的改变,高聚物表现出刚性特征。仅在适中的频率范围内,链段运动的松驰时间与应力作用时间(它被交变应力频率所决定)近于同一数量级时才表现显者的滞后现象,形成内耗。形变一应力曲线上伸缩回线构成的滞后圈封闭的面积越大,内耗越甚。轮胎等橡胶制品在交变应力下会由于内耗而导致制品温度升高,致使制品过早老化。通过设法缩小滞后圈面积可以减低这种危害。橡胶用作减震器时,则有意识地利用内耗来吸收震动波。
3.粘流性
高聚物的粘流性是一种不可逆的本体形变,与大分子链的不可逆的相对滑移有关。高聚物的熔体或浓溶液的物态属粘流态,此时大分子链基本处于紊乱状态。粘流体的流动首先是其链段的分段移动。通过链段相继移动而导致大分子的重心沿外力(如切应力)方向移动,从而形变得以实现。这一过程中,由于链段之间相互缠结,使流动产生很大的粘性内摩擦力,它包括:①由链段构象变化运动引起的不大的内摩擦所导致的粘度变化与高聚物分子量无关,而与低分子相差不多;②整个大分子链的相对位移产生塑性形变,导致很大的内摩擦,它引起的粘度很大,并依赖于分子量;③大分子链的构象变化运动,表现为链沿外力作用方向取向伸展,但链段的活动距离又被大分子链间的缠结产生的物理交联限制,使熔体的流动呈现弹粘性。高聚物的粘流态包括了上述三类形变运动。与低分子的单纯粘性流动不同,高聚物的粘流态以真实流动的塑性形变为主,夹杂了一定的非真实流动的高弹性变。
粘度是一种动态行为。高聚物的粘度是由大分子链的各种形变运动的内摩擦引起的。作为高聚物熔体的流变性特征,粘度受剪切应力的制约。当作用在熔体上的剪切应力很小时,流变速度很慢,这时大分子链段不会显出弹粘形变特征;剪切应力很大时,流变速度很大,熔体流动来不及显示弹粘形变。在这两种情况下,熔体的粘度都不依赖于外加作用力,这时的熔体流动属于牛顿粘性流动。仅在剪切应力适中时,高聚物熔体的流变过程才突出表现出弹粘形变特征,这时熔体的表观粘度随外加作用力而改变,熔体的流动属于非牛顿粘性流动。在这一适中的应力范围内,熔体流动时,大分子链段及整个大分子链间都可能有内摩擦及构象的取向变化,使液层间剪切速度的增大落后于剪切应力的增大,因而在应方剪切速度图上构成向上凸起的曲线。该曲线的斜率即为相应条件下熔体的表观粘度。显然,表观粘度值随剪切速度的提高而降低。因此,剪切速度的增大有利于改善熔体的流动性。在高剪切速度下,大分子链的构象取向程度提高,流体流层间的拖曳力减小,大分子较易发生相对滑动。这一现象对塑料的注塑成型有利,在高剪切速度下熔体的表观粘度下降较快,熔体易于注满型腔。
在开始发生取向变化时,熔体的粘度会随时间而改变。当链段及大分子链的构象均趋于与外力相适应的平衡状态时,粘度不再发生变化,这时,大分子链沿流体流动方向排列,流体处层流状态,流动的阻力减小,使熔体的粘度较低。
温度是影响高聚物流变性的另一重要外因。温度升高,大分子的各种形变运动加剧,使熔体的表观粘度降低。高聚物的链结构中含有位阻较大的基团或主链刚硬者,它们的形变活化能较大,使它们的熔体粘度在温度升高时下降得更明显。例如,聚碳酸酯、聚甲基丙烯酸甲酯等高聚物的粘度的温度效应就强于聚乙烯。
分子量、分子量分布、支化等结构因素中,能改善大分子流动性的因素都有利于降低熔体的粘度。例如,分子量分布较宽者对大分子链的运动有利,从而能增强熔体的流动性和降低粘度;降低分子量的作用与此相似。
研究高聚物的表观粘度受诸因素的制约,对优化高聚物的成型工艺极为重要。例如,在加工模塑制品时,必须根据所用树脂的性质,控制预热及模塑的时间和温度,以使树脂达到完全的粘性流动,弹粘形变充分松驰,保证了熔体在模型型腔内冷却到璃璃态时,不会有大分子来不及回复到无规线团构型就被就地“冻结”,从而避免了应力在材料内的积累。这种应力使制品易于开裂。在热塑性塑料挤出工艺中,如果使用过大的挤出压力或过快的挤出速度,都会使熔体的流动失稳,发生湍流、涡流等无规流动现象,在挤出物中出现不正常的扭曲、表面粗糙、疙瘩等,严重时导致不规则断裂。这些现象统称为“熔体破坏”。
当熔体温度接近粘流温度时,高聚物对强迫高速流动表现出明显的受压回弹行为。例如,高聚物熔体在压力下被从模口强行挤出时,会在模口外发生膨胀,挤出物的截面积大于模口的截面积。这种现象与熔体破坏现象都来自高聚物熔体的弹粘性。高聚物熔体的弹粘性以粘流性为主,受压回弹性为次。
4.虎克弹性
虎克弹性出现于玻璃态的非晶高聚物中,此时高聚物的力学性质与无机玻璃相似,处于普弹性状态。在玻璃态下,不发生大尺寸的分子运动,链段活动受到很大限制,仅有一些原子和小的基团能克服次价键力的限制而发生局部的微弱热运动可能表现为键长的伸缩形变及键角的改变,多数属于原子围绕平衡位置所作的短程振动。
在虎克弹性区域内,高聚物出现最大模量值(约为105~5.5N/cm2);温度升高时模量值的改变很小,高聚物刚硬;受力时的形变服从于虎克定律,并能在极短时间内达到平衡;外力作用所能引起的形变值很小;体积膨胀系数较小。
在玻璃化转变温度附近,非晶态高聚物的模量下降很快,虎克定律不再适用,形变开始带有不可逆性。在玻璃化转变温度以上,大分子内的较长链段的扩散运动得以进行,此时所需要的自由体积大于玻璃态下仅需满足原子的短程振动所必需的值,因而表现为T_以上的体积膨胀系数增大。
5.结晶高聚物的流变学性质的复杂性
非晶态高聚物是各向同性的,它们总带有不同程度的均一性,这就保证了在一定范围内应力的分布是均匀的。这种范围有时局限在小区域内,有时则可到整个体系。结晶高聚物由较大的晶粒聚集在一起,晶粒的空间分布方向的不均一性使结晶高聚物具有各向异性,受力时必然会在某些局部出现应力集中现象。实际上,结晶高聚物可视作无数个有序度及几何尺寸差异都很大的微区的集合。按两相模型,在集合中包括了完全有序的晶粒及完全非晶态的微粒的极端情况。应力变化引起结晶的熔体或生长,也会改变这些区域的大小及有序度。由于晶态与非晶态大分子的流变性质存在差异,同类高聚物的力学性质会随结晶度的不同而异。
增塑聚氯乙烯、弹性聚酰胺等高聚物的结晶度仅为5%~10%。它们的力学行为类似于轻度交联的非晶态高聚物,它们结构内的结晶区域对流变性的影响与交联的作用相类似。它们的玻璃态与橡胶态间的转变区域的温度范围较宽。结晶区域对大分子链的整体运动和链间滑移都有阻滞作用,该作用对时间来说是稳定的,对温度升高却不稳定。低结晶度高聚物的粘弹性质与非晶态高聚物极为相像。
结晶度为20%~60%的高聚物(如低密度聚乙烯),在伸长率小于1%、温度远低于Tm值、外力作用时间不太长时,它们的力学行为与低结晶度的高聚物相似。在高度延伸情况下,它们则呈现应力松驰及冷流现象,同时伴有结晶形态的改变。该区域内高聚物的流变学、力学行为与结构的关系还不清楚,尚缺乏适当的理论解释。在Tg以上,具有中等结晶度的高聚物表现出似皮革状特征;在略低于Tg时,则出现似角质状特征。这两种情况下它们都有良好的抗冲击性能。以抗冲击性为代表的韧性甚至可保留到远低于Ts温度下。韧性的真实结构特片还不很清楚。在Tg到Tm之间的大部分温度区间及略低于TR的场合,中等结晶度高聚物的力学性能基本保持不变,这种独特性能在机械及工程上都十分重要。
结晶度非常高(>70%)时,高聚物的模量达到接近于晶区的高模量值,同时表现出变脆的趋势,应变不大时材料就易破坏。较为典型的是高密度聚乙烯。在达到或略高过屈服应力点处,高密度聚乙烯就会发生拉伸破坏,同时伴随有畸变或形变,因此高密度聚乙烯不适于冷拉伸。通过与金属的粘弹行为比较,估计它的畸变或形变发生在滑移界面或位错的晶面上。
在远低于玻璃化转变温度下,高聚物内基本上不存在分子运动。材料的行为更像一个坚硬的玻璃状固体,与结晶的存在与否关系不大。例如,无规与全同立构聚苯乙烯的性能在室温时就极为相似。
在Tm以上,结晶度的大小对粘弹熔体的性能没有什么影响。
三、高聚物的机械性能
1.高分子材料的断裂
高分子材料的断裂行为很复杂。按照破坏的条件划分,除拉伸断裂外,还有冲击断裂、静态的蠕变断裂、动态疲劳、磨损、环境应力开裂等不同方式。描述不同破坏方式的理论也各不相同。
以裂纹为基础的宏观断裂理论认为,玻璃态高聚物在应力作用下特有一种沿拉伸方向的塑性形变方式,它与切变延性相竞争,此时材料在横向上并无收缩,因而形成非断裂性损坏。透明的热塑性塑料在损伤区会失去光学透明性;某些损伤区会出现双折射性,还会反射或散射入射光;在对着光时可以在材料内见到一些闪闪发亮的细丝般的裂纹,际为“银纹”。银纹是材料内发生高度形变且亚微观损伤很多的细带。大分子链在细带内平行于应力方向排列。它们被拉成取向的细丝或薄片,彼此间形成了互相连接的、密度较低的裂纹。与裂缝不同,裂纹区的密度虽然低于本体密度,但却不为零;即使裂纹较多时,仍可维持无裂纹状态的强度的一半以上。裂纹不断发展会导致材料破坏。
高分子量的玻璃态高聚物在受到拉伸应力时,有三类主要应变形式:(1)均匀拉伸;(2)通过形成细颈的方式被拉伸;(3)以薄层延伸方式形成银纹,最后裂缝在材料的银纹区域扩展,形成脆性断裂。可以把银纹看作是能在任何取向的硬性材料中完全自然发牛的一类过程,它常在拉伸应力使材料的应变超过临界值时形成。在压缩应力下不发生银纹。即使在拉仲形变下,只需施加足够的流体静压也能阻止银纹发展。银纹总先在应力集中点发生。作为适当应变条件下的塑性不稳定性,银纹应力也表现出粘弹性特征,它随温度的升高和应变速率的减小而减小。
断裂的分子理论用于解释共价键断裂的微观过程。一个经受应力的大分子链可机械断裂为两个很活泼的链端自由基。每个链端自由基均能与邻近的大分子链作用而生成链内自由基。链内自由基很快使大分子链断裂成一个惰性端基和另一个链端自由基。分子链断裂持续扩展的结果,形成亚微裂缝,积累到一定数量后,许多亚微裂缝并集成较大裂缝,最后导致材料破裂。
2.高聚物的重要机械性能概述
高聚物材料的弹性模量依赖于分子结构、结晶性、大分子链的柔顺性等因素。凡属分子量较大、极性较大、取向程度较高、结晶度较大、交联度较高、柔顺性较低的高聚物,都有较高的弹性模量。同一类高聚物的弹性模量随温度、作用力速度及结晶度不同而有很大变化。例如,高密度的结晶聚乙烯的弹性模量达828MPa;具有分支的轻度结晶的聚乙烯则仅为172MPa。刚性的热固性高聚物的弹性模量比热塑性高聚物高出1~2个数量级。
高聚物材料的其它机械性能与弹性模量间有一定的对应关系。弹性模量较大的高聚物的抗冲击强度较小,但硬度、挠曲强度、抗压强度较大。
非晶态高聚物在玻璃态下通常是硬而脆的,具有中等的抗拉强度。结晶性高聚物较为坚韧,其机械强度取决于定向晶体的主价键变形。由于线型大分子卷曲缠绕,在外力作用下大分子链可以拉长、伸直,并能通过链段间相互滑动而排列成行(结晶),加大分子间的内聚力,从而提高其抗张强度。尼龙、聚丙烯酸酯、聚碳酸酯等热塑性高聚物内具有使碳链僵化的基团,因而可以得到很高的抗张强度(62~69MPa),高于简单聚烯烃。热固型高聚物的机械强度取决于高分子的主价力,适度的交联可以加固分子结构,外力作用下大分子间不易发生相对滑动,而是共同承担外力,从而具有较高的抗张强度。但过度的交联又使大分子链不易伸直,反而妨碍承受外力的均匀性,又会降低抗张强度。随交联度增大,硬度和耐蠕变性也相应增大,但伸长率降低。
抗冲击强度(又名冲击韧性)是在高速冲击条件下的耐断裂性。它是高分子材料的重要使用性能,其大小为发生冲击断裂时单位断裂面积所吸收的能量。快速拉伸试验得到的应力一应变曲线下面的面积是材料韧性的一种粗略量度。冲击韧性取决于冲击的瞬时应力能否迅速分散。如果冲击作用力急速,链段来不及作松驰运动以传递、分散应力,则会在材料内部最薄弱点上出现应力集中,此时决定材料是否断裂的主要决定因素是该部位的屈服应力与发生脆性破裂时的抗张强度(即脆性强度)的相对大小。如果屈服应力或强度比脆性强度低,那么就开始塑性流动,在抗拉试验中出现屈服点,高聚物表现为韧性;如果脆性强度较低,则冲击时发生脆性断裂。因此,能提高链的柔顺性的结构因素就有利于提高高聚物的抗冲击强度。在玻璃化温度以下使用的非晶态高聚物的链段不能运动,因而冲击强度一般不高,其大小依赖于重均分子量。结晶度适中的高聚物在略高于TR时就有高冲击强度。提高结晶度,特别是增大球晶的尺寸,则会降低冲击强度。热固性高聚物的冲击强度通常低于热塑性高聚物,且在较大范围内随温度变化很小。总体说来,高聚物大分子链的柔顺性特征使高聚物的冲击强度优于其它非金属材料,但仍比金属小两个数量级。除结构因素外,高聚物的冲击强度还依赖于温度。在玻璃化转变温度附近,玻璃态高聚物的冲击强度随温度急剧上升。显然,提高材料的强度(例如纤维增强)或改善大分子链柔顺性(例如橡胶增强塑料),都可以提高高聚物的抗冲击强度。
在高分子材料的伸长率方面,热塑性材料中,延性的聚乙烯、聚丙烯的伸长率明显高于脆性的聚苯乙烯、聚甲基丙烯酸甲酯等。通过在苯乙烯中共混入橡胶或合成含苯乙烯的共聚物(如ABS)都能使其塑性明显改善。热塑性材料的伸长率在玻璃化温度以下明显降低。外力高速作用将大大降低伸长率。热固性材料的网状结构阻止了大分子链的伸直,在所有温度下它们的伸长率都很低。
作为材料表面的长期性质,塑料的摩擦系数总的说来较高(大约为0.2~0.6)。摩擦力是多种复杂因素的总和,它包括了由力学阻尼引起的内摩擦及发生在直接接触表面结合点处的剪切作用导致的表面切摩擦等作用。前者又被称作分子粘接作用,它对于以汽车轮胎的滚动摩擦为代表的循环应力作用过程最为重要;后者又可称作机械粘结作用。机械粘结作用与表面的亚微观粗糙性有关。两个面接触时,仅有少数点发生实际接触。当外力迫使一个面在另一个面上滑动时,接触点上会产生很大的局部作用力,往往发生局部微区的塑性形变,使结合点“焊接”到一起,因而,即使在软材料的下方也会出现剪切摩擦。外力强制下的表面滑移会引起瞬时结合的周期性断裂。表面切摩擦的极端形式为硬材料与软材料的接触,此时硬材料的凸出部位犁进软材料表层。高聚物的硬度普遍较低,因而整个摩擦力中这种“犁入”的作用可能很重要,同时“犁入”作用将产生强烈的磨蚀和磨耗。总的说来磨蚀与摩擦密切相关。高聚物的磨蚀正比于高聚物的抗张强度。
部分塑料有独特的摩擦性能。聚对苯二甲酸乙二酯、尼龙、高密度聚乙烯等有较低的磨耗因子。聚四氟乙烯有极低的摩擦系数。尼龙、聚甲醛、聚四氟乙烯等有良好的自润滑性能,即在无润滑的摩擦条件下仍有较低的摩擦系数及耐磨性能。以聚四氟乙烯为例,由于氟原子有效地屏蔽了主链碳原子,使大分子间的相互引力小,表面分子对其它分子的吸引力也很小,因而具有非常小的摩擦系数,在高速高负荷的条件下可低至0.01。另外,聚四氟乙烯的摩擦系数不随温度而变化,可从超低温保持到其熔点。聚四氟乙烯的自润滑性必须在对磨材料表面具有合适的光洁度时才能体现。通常当对磨材料表面不平度在0.1~0.4μm时,聚四氟乙烯的磨耗最小。这种现象的实质是:在转动或滑动时,具有合适的不平度的对磨表面因磨耗聚四氟乙烯而被覆上一层薄膜,使两类不同材料的对磨变为低硬度的聚四氟乙烯的自身磨擦,此时它的自润滑性充分发挥,摩擦系数极低,使其磨耗量降至很小。
3.改善高聚物机械性能的途径
高聚物的机械性能可通过多种工艺方法加以改进,其中最主要的是形成共混高聚物及复合材料。共混高聚物是由数种或两种以上化学结构不同的高聚物组成的混合物体系,复合材料则是高聚物和非高聚物(主要是无机材料)构成的物料体系。
(1)复合材料——增强和填充改性
在高聚物基体中加入第二相物质,常可显著改善材料原有的某些力学性能,如强度、耐磨耗性、耐热性能等。
纤维具有极高的抗张强度和模量。在高聚物基体中有效地使用高强度纤维(例如玻璃纤维石棉纤维、碳纤维、硼纤维、合成纤维)及近年发展起来的无机物晶须等,可制得强度高于块状高聚物材料的复合材料,称为增强。
工程塑料经增强后,静态强度(如抗张强度,抗弯强度等)能提高2~3倍,动态强度(如疲劳强度、冲击强度、耐蠕变性、刚性等)能提高2~5倍,热变形温度可提高10℃~200℃,线膨胀系数和成型收缩率能降低1/4~1/2趿水率能下降10%~20%。增强改性的不足之处是制品接缝强度和光泽度有所降低透明度受到影响容易出现表面粗化现象,有些品种的机械性能、成型收缩率及线膨胀系数等会出现不同程度的各向异性。
在纤维增强复合材料中有三种基本单元:纤维、高聚物基体及纤维基体界面。纤维赋予复合材料以高强度和高模量,主要用来承受应力并抑制裂纹的扩展,提供在应力或负荷作用下的抗破裂和抗挠曲性能。高聚物基体(树脂)作为粘结孤立纤维并将应力传递和分配到各纤维个体上的基材,使多根纤维作为一个整体来抵抗负荷下可能产生的破坏和变形,同时保护纤维免于磨损并与环境介质相隔离。纤维基体界面是决定复合材料在使用过程中能以何种程度发挥并维持其潜在性能的关键因素。通常在界面及界面附近,应力集中最大,这里可能成为复合材料过早破坏的场所。纤维基体界面必须有适当的化学和物理特性以促使负荷从基体转移到增强纤维上。
使用偶联剂可改善界面状况。硅烷是最常使用的一类偶联剂。硅烷分子具有通式为RnSiX4n的结构,其中x为可水解基团,能与玻璃纤维表面键合,而硅烷分子又通过有机基团R与高聚物基体形成共价键桥从而以硅烷分子作为中面过渡,把玻璃纤维与高分子链有机地结合在一起,显著增强了界面粘合力,使复合材料获得较好的物理性能。例如经偶联剂处理后的玻璃纤维增强的聚甲醛,其伸长率、抗张强度和抗弯强度可提高50%以上,缺口冲击强度可提高1倍以上,并且解决了单纯用玻璃纤维增强时聚甲醛的各向异性及玻璃纤维的定向问题。
以粉状物料填充到高聚物中,多数情况下能改进高聚物材料的性能粉状填料的加入通常不是单纯的物理混合。在填料与聚合物之间存在着次价力,这种次价力虽然很弱,但是具有加合性,从而能以填料粒子为中心,构成多条大分子链的物理交联。其中一条链受到应力时,能通过交联点把力分散到其它链上。大部分填充剂能提高工程塑料的机械强度,最突出的影响是使复合材料变硬,降低材料的线膨胀系数和成型收缩率,使工程塑料能在更宽的温度范围内工作。填料的加入能降低高聚物的结晶倾向和溶解度,使高聚物熔体粘度增大。填料改性往往会降低高聚物材料的抗张强度、抗挠曲强度及抗冲击强度,使材料的脆性增大。
填料颗粒事先经偶联剂或粘结剂处理后,能改善填料基体界面的接合能力。起到一定的增强作用。
(2)共混
以物理或化学的方法将不同高聚物混合,所制得的高聚物的混合体系被形象化地称为“高分子合金”。共混高聚物的特点是能改进某一方面的性能而不显著降低其它性能。两种高聚物组成的物料体系可能是均相的,也可能是非均相的。均相体系的共混聚合物主要能提高熔体的流动性,以改进加工性能。非均相体系则主要能改进共混产物的柔韧性从而提高抗冲击强度。例如,聚苯醚虽具有优异性能,但熔融流动性差,成型加工困难,限制了它的发展。然而在其中渗混一定数量的高抗冲聚苯乙烯后,除了耐热性比聚苯醚稍低外,其它优异性能均能保持,使共混产物的成型加工性能大大改善。少量的聚乙烯与聚碳酸酯共混后,能降低熔体粘度,改进聚碳酸脂的成型加工性能,同时还能进一步改善其冲击强度、抗张强度、断裂伸长率等,而热变形温度仍可维持其原有水平。工业上极为重要的非均相体系的应用是橡胶对塑料进行改性以提高某些塑料的柔韧性。例如橡胶与聚苯乙烯共混。
制备共混产物通常使用的物理混合方法,包括机械混合(即采用挤出机在各单元组份均处于熔融状态下进行混合)以及溶液浇铸和乳液混合等。化学混合法是一种较新的方法,它包括交联网络及溶液接枝等手段。其中,用交联网格法制备的高分子合金具有优异性能。它是用一种能够钻入某交联聚合物网络的单体使前者溶胀后,然后使单体就地聚合。它也可以用两种未曾交联的高分子胶乳进行混合,再加入固化剂交联而得。由于交联网络产物内,相域的微观结构很微小,相互构成牢固的结合,因此如果一种交联聚合物是弹性体,另一种是塑性体,则它们将在共混产物中表现出协同作用,所获得的共混产物可作为增强的弹性体、高抗冲塑料或消除噪音及吸震用材料。
广义的“共混高聚物”还包括了嵌段共聚物及接枝共聚物。工业上把未交联的丁苯橡胶溶于苯乙烯单体中,然后进行聚合,这样得到的产物混合较为均匀。由于在丁苯橡胶的大分子链上接出许多聚苯乙烯短链,使改性产物的抗冲击强度比聚苯乙烯高7倍。ABS树脂作为三元接枝共聚产物,有远高于聚苯乙烯的抗冲击性能及耐热、耐溶剂性能。聚氨酯橡胶作为二异氰酸酯与含端羟基的聚酯或聚醚的嵌段共聚产物,有优良的耐磨性及抗冲击性能。它可以用热塑性塑料的成型方法直接加工成橡胶制品,不需要硫化步骤。
(3)交联、取向及其它方法
适度交联可提高高分子材料的机械强度。例如经高能射线照射后,聚乙烯的大分子链间产生交联键,可以大大提高聚乙烯的拉伸强度、硬度、抗挠曲强度及使用温度等性能,断裂伸长率及溶解度降低。聚乙烯也可用有机过氧化物作为引发剂使大分子间产生交联键。将交联剂及其它助剂与聚乙烯一起混炼,其混炼温度应高于聚乙烯的熔融温度,低于交联剂的分解温度。在成型时提高熔体温度,使交联剂分解,引发链间交联。
取向对高聚物强度的提高作用已如前述。它主要能改善高聚物的抗张强度,并显著提高其断裂伸长率及抗冲击强度。
采用异相成晶技术,能使结晶性高聚物生成微晶结构,结晶速度加快,产物的晶粒尺寸减小,结晶度提高且均匀,具有较高的强度。该技术在成晶时从外部加入细微粉状的无机物或有机物作为成核剂或晶种。经常使用的加入物是与待结晶高聚物有相同的内聚能密度而其熔点又较高的物质。
四、高聚物的环境性质
高聚物的环境性质包括热稳定性、化学稳定性及其它物理稳定性(光、电、高能辐射等)、生物稳定性、可燃性及耐有机溶剂的性质。
高分子材料在加工、贮存及使用过程中,由于多种因素的影响,逐渐失去原有的优异性能,以至最终丧失使用价值,这一现象称作“老化”。高分子材料随种类及老化条件的不同,表现多种多样的老化现象,如塑料薄膜制品的变色、脆化、破裂,涂料层的变化、粉化、起泡、龟裂、脱落,橡胶制品的发粘、变软、变硬、脆化、龟裂、发霉等以及物理机械性能的恶化(如强度降低、弹性消失、绝缘性能下降等)。老化现象是不可逆的,它是高聚物的主要缺点之一。
高分子材料在热、氧等外界因素作用下,本身结构发生了变化,导致大分子链的断裂(称为降解或裂解)、交联或其它结构改变,在外观上表现为老化。在高聚物的降解过程中,大分子链的断裂使分子量下降,有时甚至分解成为单体,使材料发粘变软,丧失原有的机械强度。大分子链或断链间的交联、支化或环化使材料变硬、变脆,丧失弹性。一般认为,在这类过程中发生的化学变化主要是游离基的反应过程。高聚物在氧(臭氧的怍用更甚)、光、热及射线的作用下,被引发出多种游离基,这些高活泼性的游离基促使主链断裂,再进一步出现一系列的结构改变,发生降解或交联反应,最终使高聚物的力学性质(强度、延伸性、回弹性等)变坏。氧化往往是使高分子材料老化的主要原因。辐射总是加速氧化反应。高分子材料中含有的易分解为游离基的引发剂或某些过渡元素(如Fe,Cu,Mn,Ni离子)皆能促使氧化降解的进行。
高聚物吸收氧进行反应时则发生氧化。其吸氧速度依高聚物的分子结构而异,因此可以用测定吸氧速度来评价高聚物易氧化的程度。基于吸氧速度的测定结果,认为具有双键的高聚物最容易被氧化,例如天然橡胶及聚丁二烯橡胶。其它吸氧速度快的高聚物有聚丙烯、聚乙烯、尼龙、聚氯乙烯、聚乙烯醇、聚丙烯酸酯类。吸氧速度非常慢的高聚物有聚酯橡胶、聚甲基丙烯酸甲酯、聚苯乙烯、硅橡胶等。
聚双烯类橡胶的大分子主链上含有极多双键,双键a—碳上的C—H是弱键,极易被活化和氧化。吸小浓度的臭氧也会引起橡胶和臭氧化,即使在低温、阴暗处臭氧仍能与多种橡胶作用。臭氧通常先在橡胶表面层生成臭氧化薄膜,然后薄膜龟裂,使臭氧老化向制品纵深发展。延缓橡胶的臭氧化过程的方法包括:采用物理抗臭氧剂(在配方中加入石蜡或将制品表面喷蜡,以隔离橡胶与臭氧的接触),加入化学抗臭氧剂(某些对苯二胺衍生物除开具有抗臭氧效能外,还有抗氧及抗疲劳老比性),混入耐臭氧性高的胶类(如在不耐臭氧的丁腈橡胶中混入氯丁橡胶),加入陶士或碳黑等填充剂等。通过提高结晶度来降低链段的活动性,也可以达到抑制臭氧龟裂生长的目的。
高聚物的热稳定性可视为在热影响下其使用性质(如强度、绝缘性、结构完整性等)的稳定性或保持性。高聚物的熔点或分解温度总是构成热稳定性的上限,“使用温度”总要代些。升温至使高聚物的化学键离解时,发生大分子的热裂解。聚甲基丙烯酸甲酯、聚四氟乙烯等在热裂解时还原为其单体,称为键式裂解。另一些高聚物(如聚乙烯、聚丙烯等)则主要形成分子量相对较大的碎片,称为无规降解。
许多高聚物的耐热能力主要决定于最弱键的强度。因此,键节上原子或基团间的键结合能的大小对耐热性影响很大,应尽量避免在主链结构上出现低裂解能的键。可采用的提高高聚物耐热能力的办法包括:在主键上引入高键能的原子或基团,引入共轭双键体系,构成环状分子链或梯形结构,提高效联密度,提高高聚物的结晶度,增强等。例如,热固性塑料固化后的耐热老化性能就比交联固化前好。
在有机液体作用下,高聚物可能发生溶解、溶胀、环境应力开裂、环境银纹等,从而导致高分子材料使用性质的破坏,在各种有机液体作用下,非晶态及半结晶材料都会出现应力开裂和银纹现象,其破坏应力远低于抗张强度及在空气中出现银纹的临界应力,尤其在玻璃状高聚物中溶剂银纹可视为材料内在的弱点。半结晶材料的问题稍轻。溶解、溶胀和溶剂开裂是紧密联系的现象。有机溶剂在开始时是使高聚物溶胀,其结果降低了,使在一定温度下发生塑性流动的应力减小。溶解或开裂谁占优势则取决于溶剂渗透的速率和裂缝形成的速率的相对大小。
不带极性基团的高聚物耐化学药品侵蚀的能力较高,尤基主链键能高的,更不易在化学药品侵蚀下断链,有更好的耐腐蚀性。聚四氟乙烯的优异耐腐蚀能力来自其分子结构,氟原子在碳链周围组成一个保护层,屏蔽了碳链,使其不易被侵蚀。在分子结构相同的情况下,分子量大或结晶度高的高聚物的耐腐蚀性较好。
高分子本身的化学结构和物理结构是高分子材料耐老化性能好坏的基本因素。例如,具有硅氧主链结构的高分子就比具有碳碳主链结构的高分子的耐老化性能好,这是因为硅氧键结合的键能比较大,因此需要外界有较大的能量才能使硅氧键断裂。聚丙烯的耐氧、光、热老化性能较差,因为在聚丙烯分子结构中含有大量叔碳原子,叔碳上的氢易被氧化。其它结构因素如聚集状态、结晶度、立体构型的规整性、取向性、交联度、键的不饱和程度、分子量大小及分子量分布等,都会影响高分子材料的耐老化性能。
对高分子材料的老化加以防护,虽然不能消除老化现象,但却可以延续这一过程,从而抑制老化的破坏作用。随老化机制的不同,延缓老化的方法也不同。高聚物的防老化总括起来有以下途径:
1.添加各种稳定剂,如抗氧、抗臭氧剂、紫外线吸收剂等。
2.表面防护,如表面涂层、表面保护膜。
3.改进高聚物结构,以减少各结构层次的弱点,如各种改性方法。
4.改进聚合条件和方法,例如采用高纯度单体、改进聚合工艺、改进后处理工艺、减少高聚物中残留的催化剂等。
5.改进高聚物的加工成型工艺。以减小强烈环境因素的作用时间及强度。
6.改进高聚物的使用方法,例如避免不必要的阳光曝晒及烘烤等。
五、高聚物的其它性能
高聚物结构中没有可自由移动的电子及离子,所以导电能力很低,大多是优良的绝缘材料。高聚物的介电性能与高聚物的结构、组成原子的电性能、分子的对称性、分子内极性基团的位置等内部化学构成因素有关。非极性烃类及具有电子平衡结构的高聚物受电场的影响极小,是优良的高频电绝缘材料,例如聚四氟乙烯及聚乙烯。高聚物内夹杂的杂质离子的运动会引起微量电导,制品表面附有外来的导电物质(如潮气)会降低它们的表面电阻率,都会削弱高聚物的绝缘能力。但表面电阻率的下降,能防止高聚物由于摩擦而发生的静电现象,对于合成纤维制品、电影胶片等的应用又是有利的。
持续升高施加在高分子材料上的电压,将会达到一个极限,其时由于电介质的物理击穿引起电阻的急剧下降。单位厚度的绝缘材料上承受的击穿电压称为介电强度(或击穿强度)。高分子材料的电击穿现象较为复杂,其过程可能是电击穿或热击穿。前者是因为介质在外加电场作用下发生电荷位移,直至使高分子电离,生成的离子或电子以撞击方式连锁击发出新的电子,最终导致电子积累达到某一临界点,出现击穿;后者则是由于介质在电场中由于导电损耗、介质损耗等引起发热,温度升高又使电导进一步增大,直到介质产生过高漏导或介质局部碳化,使材料的绝缘性能破坏。高聚物的电阻率随温度升高而下降,使击穿强度也随之下降。高弹态时的电阻率与击穿强度要比玻璃态时低,因此提高高聚物的玻璃化转变温度及耐热性是提高击穿强度的途径。
刚性的非晶态高聚物往往有较好的透明性,且对可见光的作用呈各向同性。为保持这种性质,在塑料成型时应避免应变取向及留下流动痕迹。同种高聚物,处于结晶态时的密度总高于非晶态。由于折射率总是与密度成正比关系,使得部分结晶的高聚物表现为由具有不同折光指数的分散区域构成,对可见光而言,这种差异导致散射。如果这些区域(微晶或片晶、球晶)的尺寸大小与可见光波长相当,则散射就会很大。因而经接枝共聚的抗冲击型高聚物及结晶态高聚物大多表现为半透明或不透明,它们的透明度随晶体尺寸的减小而增加。如果控制结晶过程在快速冷却条件下进行,能使所得晶体变细。当生成的晶体粒度低于可见光波长时,高聚物转呈透明。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。











