



实验二十 GFP和HSF4的融合蛋白在HeLa细胞中的定位观察
一、实验原理
绿色荧光蛋白(Green fluorescent protein,GFP)由238个氨基酸组成,分子量为26.9ku,是从维多利亚多管水母中提取的能够自发绿色荧光的蛋白质。由于它的自发荧光具有荧光强度强、不易淬灭的特性,因此,常作为一种理想的报告分子,在分子生物学和细胞生物学领域得到广泛应用,在检测蛋白质表达、研究蛋白质之间相互作用以及构象变化、蛋白质和细胞荧光示踪中,起到了重要的作用。EGFP是一种优化的突变型GFP,使其产生的荧光较普通GFP强35倍,大大增强了其报告基因的敏感度。EGFP与其他蛋白的融合表达已有很多成功的例子,而且其N及C端均可融合,并不影响其发光。
本实验利用基因重组技术将HSF4蛋白(一种热休克转录因子)插入到含GFP的哺乳动物表达质粒pEGFP-C1中,再将重组质粒转染到HeLa细胞中进行表达,最后通过观察、比较、分析绿色荧光在细胞中的分布来判断HSF4蛋白在细胞中的定位情况。
二、实验设计
(一)GFP和HSF4的融合蛋白表达载体的构建
1.PCR扩增目的基因片段:
(1)引物设计:
HSF4b cDNA全序列如下:
atgcaggaagcgccagctgcgctgcccacggagccaggccccagccccgtgcctgccttcctcgg
caagctatgggcgctggtgggggacccaggcacagaccacctgatccgctggagcccgagcggg
accagtttcctcgtaagcgaccagagccgtttcgccaaggaagtgctgccccagtatttcaagcat
agcaacatggcgagcttcgtgcgccaactcaacatgtacggttttcggaaggtggtgagcatcgag
cagggcggcctgcttaggccggagcgcgaccacgtcgagttccagcacccgagcttcgtgcgcgg
ccgcgagcagctactggagcgcgtgcggcgcaaggtgcccgcgctgcgcggcgacgacggcc
gctggcgcccggaggacctgggtcgactactgggcgaggtgcaggctttgcggggagtgcagga
gagcaccgaggcgcggctgcgggagctcaggcagcagaacgagatcttgtggcgggaggtggt
gacacttcggcagagccacggtcagcagcaccgggtcattggcaagctgatccagtgtctctttggg
ccacttcaggcggggccgagcaatgcaggaggcaagagaaagctgtccctgatgctggatgagg
ggagctcatgcccaacacctgccaagttcaacacctgccctctacctggtgcccttctgcaggacc
cctacttcatccagtcgcctctcccagagacaaatttgggccttagccctcacagggccaggggcc
ccatcatctctgacatcccagaagactctccatcccctgaggggaccaggctttctccctccagtgat
ggcaggagggagaagggcctggcactgctcaaagaagagccggccagtccagggggggat
ggcgaggccgggctggccctggccccaaacgagtgtgacttctgcgtgacagcccccccgcca
ctgcctgtggctgtggtgcaggccatcctggaagggaaagggagcttcagccccgaggggccca
ggaatgcccaacagcctgaaccaggggatcccagggagatacctgacagggggcctctgggcc
tggaaagcggggacaggagcccagagagtctgctgcctccgatgctgcttcagccccctcaag
aaagtgtggaacctgcagggcctctagatgtgctgggccccagtctccaagggcgagaatgga
ccctgatggacttggacatggagctgtccttgatgcagcccttggttccagagcggggtgagcctg
agctggcggtcaaggggttaaattctccaagcccagggaaggaccccacgctcggggcccca
ctcctgctggatgtccaggcggccttgggaggcccagccctgggcctgcctggggctttaaccattt
atagcactcctgagagccggactgcctcctacttgggcccggaagccagtccctccccctaa
根据要求供需设计2条引物,分别为N端引物,C端引物。
上游引物(N端引物)与cDNA相同,下游引物(C端引物)与cDNA反向互补。每条引物包含三个部分,分别为酶切位点保护碱基、酶切位点以及与模板配对部分。上游引物引入XhoⅠ酶切位点(识别序列为ctcgag),下游引物引入HindⅢ酶切位点(识别序列为aagctt)。
(2)PCR扩增(实验原理见实验九)。
①PCR退火温度的确定:设计引物时应尽量使上下游引物的Tm值相近,最好不超过2℃,PCR退火温度应以Tm值最低的引物为准。Tm=2×(A+T)+4×(G+C),计算时不计入酶切位点以及保护碱基的ATGC数目。
退火温度应在52℃左右并通过实际实验情况进行调整。
②PCR的反应体系:使用KOD-Plus-DNA多聚酶(高保真DNA聚合酶)进行PCR扩增,以确保PCR的保真度,这里以TOYOBO公司的KOD-plus高保真PCR试剂盒为例来说明。反应体系为50μL,其中含有酶1μL,质粒模板1~50ng,上下游引物(10μmol/L)各1μL。50μL反应体系如下
用于KOD-Plus-的10×PCR缓冲液 5μL
2mmol/L dNTPs 5μL
25mmol/L MgSO4 2μL
引物混合液(每种10μM) 2μL
模板 DNA ≥1μL
KOD-Plus-DNA多聚酶 1μL
经过高压蒸汽灭菌处理的蒸馏水 补至50μL
在根本没有检测到某个目标物的扩增时,应考虑提高反应混合物中的MgSO4的浓度(从1mmol/L提高到1.2mmol/L)。当出现拖带时,应考虑降低反应混合物中的MgSO4的浓度(从1mmol/L降低到0.8mmol/L)。
③PCR循环(参照实验九)。
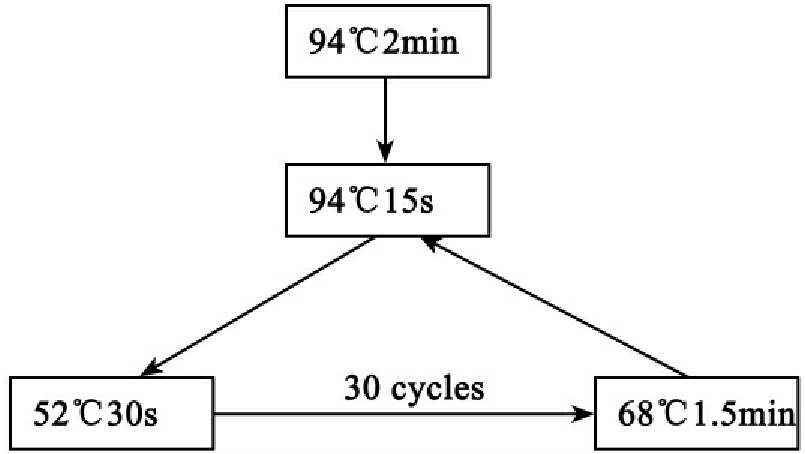
三阶段循环条件为:
变性:94℃、15秒;结合:(Tm-5)℃、30秒;伸长:68℃、1分钟/kb;25到35次循环。循环如下:
④PCR产物纯化(参照实验五):按照试剂盒说明书进行纯化。
2.双酶切及胶回收(参照附录1和实验五):
20μL的酶切体系如下:(单位μL)
ddH2O 7
10×Buffer 2
PCR产物(或质粒)* 10
HindⅢ 0.5
XhoⅠ 0.5
可根据PCR产物的浓度以及酶活力调整用量,反应温度37℃,反应时间5h(可根据底物量以及酶活力调整),酶切反应完之后将体系全部进行电泳,电泳时间应足够长以便于切胶,并用试剂盒进行回收。
3.目的片段与载体的连接及转化:
(1)载体的获得(参照实验四):
将带有载体质粒的大肠杆菌接种到含有卡那霉素的液体LB培养基中,37℃,250rpm振摇培养12h后,参照实验四中的步骤1~12进行质粒抽提获得载体。
本实验使用的pEGFP-C1质粒带有卡那抗性(如图20-1),故应加入卡那霉素,浓度为100μg/mL。

图20-1 pEGFP-C1质粒结构示意图
(2)连接(参照实验六):PCR产物和质粒经双酶切及胶回收后进行连接。连接时目的片段与载体的摩尔比一般为3∶1,即

目的片段大小为1.5kb,pEGFP-C1空载体大小为4.7kb。片段浓度可通过电泳照片,用软件计算得到,也可以使用分光光度计进行测量(参照附录6)。连接体系中总的DNA含量应不超过50ng。
总体积为10μL的反应体系一般包括T4DNA Ligase,0.5μL;10×Ligase Buffer,1μL;PCR片段以及载体片段按上述比例进行加样,以ddH2O补足体积至10μL。反应条件:16℃保温16h。反应体系和反应条件可参照酶说明书。
(3)转化(参照实验七、实验八):取实验七所制备的感受态细胞100μL→加入连接产物5μL,混匀→冰浴30min→42℃热击90s,之后立即置于冰上约5min→加入200μL无抗生素的培养基,37℃下180rpm培养1.5h→将复苏培养后的菌液(100μL每平板)涂布于含有卡那霉素的平板上,37℃倒置培养12~16h。
(4)挑取单菌落及质粒抽提(参照实验四)
取灭过菌的10mL离心管(最好用15mL离心管),每管加入4mL含有卡那霉素的液体LB培养基。用灭菌的牙签挑取生长良好的单菌落接种于离心管的培养液中。37℃,250rpm振摇培养过夜(12h,培养时间不应过长)。
次日,将培养液进行质粒抽提。
沉淀物自然干燥后,加入25μL含有RNase的TE缓冲液溶解沉淀。即得质粒DNA。
抽提之后可以取2~5μL进行电泳,也可直接用于下一步的酶切验证。
(5)表达载体的双酶切验证:双酶切体系中含有HindⅢ和XhoⅠ各0.5μL,体系总体积可以是10μL,如果抽提的质粒浓度较低也可以是20μL。10μL体系如下:
10×Buffer 1μL
抽提质粒* 8μL
HindⅢ 0.5μL
XhoⅠ 0.5μL
*可根据质粒浓度以及酶活力调整用量,用ddH2O补齐至10μL。
反应温度37℃,反应时间2h。
取酶切产物5μL电泳,如果能看到切下的目的片段条带以及载体条带则说明所需克隆的片段可能已经正确插入到质粒的多克隆位点上,该质粒随后还要通过测序进一步确认序列的正确性。
(二)HeLa细胞的培养以及质粒的转染(可参考细胞生物学实验教程)
1.HeLa细胞的传代培养:细胞生长至高密度时,即须分殖至新的培养瓶中,一般稀释比例为1∶3~1∶6。
(1)实验材料:无菌磷酸生理缓冲液(PBS):
预先配置PBS(0.1mol/L,pH7.4)储存液配方(10×):80g NaCl,32.3gNa2HPO4·12H2O,4.5gNaH2PO4·2H2O,加蒸馏水定容至1000mL。储存液室温下保存一个月。工作液(0.01mol/L,pH7.4):取储存液10倍稀释后24h内使用。
trypsin-PBS:2.5% trypsin-0.01mol/L PBS。
1640培养基:GibeoBRL干粉配制,加入双抗(青霉素、链霉素100单位/mL)。
DMEM培养基(高糖):GibeoBRL干粉配制,无抗生素。
小牛血清:灭活。
Lipofectamine试剂盒。
(2)实验步骤:HeLa细胞属于附着型细胞(adherent cell),其传代培养的具体操作步骤如下:
①吸掉培养液。
②用0.01mol/L PBS洗涤细胞一至二次。
③加入trypsin-PBS溶液37℃作用数分钟,于倒立显微镜下观察,当细胞将要分离而呈现圆粒状时,于超净工作台中吸掉trypsin-PBS溶液。
④轻拍培养瓶使细胞自瓶壁脱落,加入适量之新鲜的含小牛血清1640培养基(小牛血清含量依细胞状况调整,10%~20%),以吸管上下吸放数次以打散细胞团块,混合均匀后,依稀释比例转移至新的培养瓶中,以37℃,5% CO2培养。
2.HeLa细胞的计数
计算细胞数目可用血球计数盘。血球计数盘一般有二个chambers,每个chamber中细刻9个1mm2之大正方形,其中4个角落之正方形再细刻16个小格,深度均为0.1mm。当chamber上方盖上盖玻片后,每个大正方形之体积为1mm2×0.1mm=1.0×10-4mL。使用时,计数每个大正方形内之细胞数目,乘以稀释倍数,再乘以104,即为每mL中之细胞数目。
3.构建质粒的转染:
(1)把生长至密度为104个/mL的HeLa细胞用含20%小牛血清的DMEM(高糖)传代于12孔板,培养48h。
(2)对于每孔细胞,使用50μL无血清DMEM稀释1.0μg质粒DNA。
(3)对于每孔细胞,使用50μL无血清DMEM稀释1μL至4μL Lipofect,室温混匀,室温放置2min,在5min内与质粒DNA混合。
(4)将稀释质粒DNA与稀释Lipofect混合,室温保温20min。
(5)移去12孔板中的生长培养基,替换为每孔0.5mL无血清DMEM,接将100μL质粒-Lipofect复合物加入到每孔中,摇动培养板,轻轻混匀。
(6)在CO2培养箱中37℃培养24h。
注意:本实验转染的质粒应包括重组质粒与空载质粒两种,其中空载质粒作为阴性对照。
(三)HSF4与GFP融合蛋白在HeLa细胞中的定位观察与分析
1.将上述经瞬时转染后再培养了24h以上的细胞移出培养箱,并用PBS溶液漂洗细胞3次,每次3min。
2.加入4%多聚甲醛室温下作用30min,以将细胞固定。
3.再用PBS溶液漂洗细胞3次,每次3min。
4.避光条件下,加入DAPI染液对HeLa的细胞核染色10min。
5.用PBS溶液漂洗细胞3次,每次3min。
每个样本中可留一些PBS以保持细胞湿润,这时便可用于观察。
细胞样本的观察:将细胞样本放到荧光显微镜下进行观察,操作时要根据荧光标记的情况选择不同波长的激发光及发射光,注意避免各个光谱之间的重叠,否则会因为“串光”而干扰数据。本实验用了EGFP以及DAPI两种荧光标记,根据它们的光谱(如图20-2)情况,EGFP激发光为458nm,发射光为480~520nm;DAPI激发光为364nm,发射光为420~450nm。

图20-2 EGFP(A)以及DAPI(B)的光谱图(A曲线为激发光,B曲线为发射光)
三、结果分析
1.对克隆结果的分析:重组质粒经过测序后,应从以下几个方面对之进行分析:①插入的基因片段序列是否正确?是否有不应有的碱基的插入、缺失、替换等。②插入片段两端的酶切位点以及周围序列是否正确?只有序列全部正确,符合预期才能用于下一步的转染。
2.对转染结果的分析:在荧光显微镜下进行观察时,要将每个样本的DAPI以及EGFP被激发后产生的图像进行保存,用于对转染以及定位结果的分析。
观察并比较两种质粒所表达的EGFP被激发后产生的图像,如果两种图像中绿色荧光在大多数细胞中均有分布并且有一定的亮度,证明转染效率较高,反之则较低。
3.对蛋白质定位结果的分析:观察并比较两种质粒所表达的EGFP以及DAPI被激发后产生的图像,根据DAPI的图像确定细胞核的位置,再根据相同视野下EGFP的图像即可判断GFP和HSF4的融合蛋白在细胞中的大致位置,从而对HSF4在HeLa细胞中的定位做出相应的推论。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










