



实验四 血凝素基因的扩增
一、实验目的
掌握用RT-PCR法扩增病毒基因的方法。
二、实验原理
聚合酶链式反应(polymerase chain reaction,PCR)可用来扩增位于两段已知序列之间的DNA片段。
如图4-1所示,在由DNA聚合酶催化的一系列合成反应中,使用了两段寡核苷酸作为反应的引物。在一般情况下,这两段寡核苷酸引物的序列互不相同,并分别与模板DNA两条链上的各一段序列互补,并且这两段模板序列又分别位于待扩增DNA区段的两侧。
反应时,首先在两段寡核苷酸以及4种dNTP参与下对模板DNA进行加热变性。随后,将反应混合物冷却至某一温度,这一温度可使引物与它的靶序列发生退火。此后退火引物在DNA聚合酶作用下得以延伸。如此反复进行变性、退火和DNA合成这一循环。由于一轮扩增的产物可充当下一轮扩增的模扳,所以在这一周而复始的过程中每完成一个循环,就可使目的DNA产物增加1倍。
PCR扩增技术在医学与生物学有如下用途:
(1)遗传性疾病的检测;
(2)遗传学鉴定;
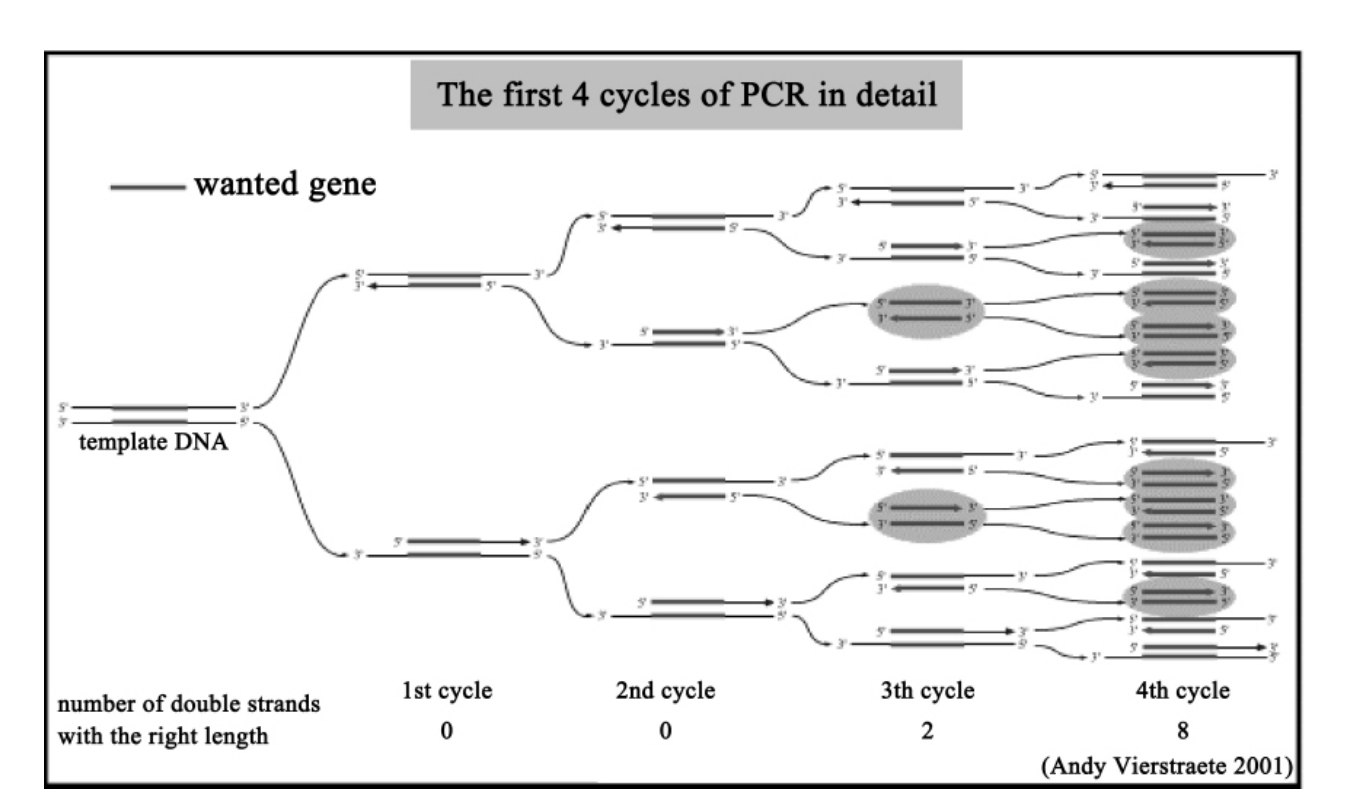
图4-1 聚合酶链式反应示意图
(3)基因突变位点的分析;
(4)通过选择性扩增特定DNA区段,生成特异性探针;
(5)由少量mRNA生成cDNA文库;
(6)染色体位移(chromosome crawling)。
三、实验材料及器械
(1)逆转录酶及其缓冲液、Taq酶及其缓冲液、dNTP、引物、无菌ddH2O。
(2)病毒RNA。
(3)tip头、Eppendorf管、PCR仪、水浴锅、微量加样器。
四、实验步骤
1.逆转录
在冰上按顺序加入以下试剂:
无RNase ddH2O 定容至5μl
RNA样品 0.05~1μg
逆转录Primer(或者随机6聚体引物) 1μl
总体积为 5μl
(2)将以上试剂混匀,在72℃下保温2min,立即置冰浴2min。
(3)在冰上按顺序继续加入以下试剂:
5×第一链反应缓冲液 2μl
DTT (20mmol/L) 1μl
dNTP (10mmol/L each) 1μl
SuperScriptTMⅡReverse Transcriptase 1μl(200unit/μl)
(4)混匀,在42℃下保温约1h,置于冰上终止第一链合成反应,即得cDNA样品。94℃5min以灭活逆转录酶。-20℃保存备用。
2.PCR
在冰上按顺序加入以下试剂进行PCR反应:
无核酶ddH2O 80μl
10×cDNA PCR缓冲液 10μl
dNTP mix (10mmol/L each) 2μl
primer 1 and2 4μl
cDNA样品 2μl
混匀,按以下程序进行PCR反应。
94℃5min;
94℃30s,50℃60s,72℃30s,30个循环;
72℃延伸5min。
取10μlPCR产物并加入2μl6×上样缓冲液于1.0%琼脂糖凝胶上电泳,在紫外灯下观察并照相记录(图4-2,电泳后可在紫外灯下可见一条约1.7kb的条带)。
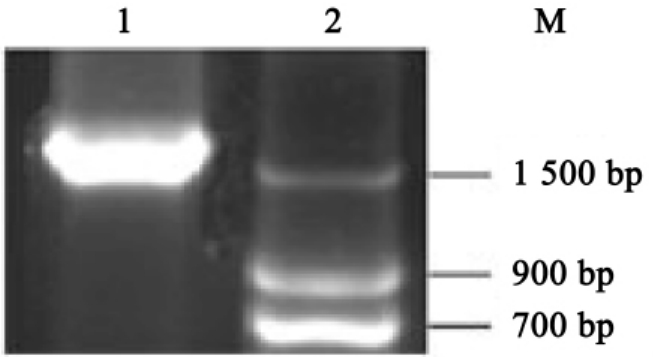
图4-2 HA PCR结果
五、方法评注
1.背景介绍
多聚酶链式反应1971年由Khorana等首先提出,他们认为可在体外经DNA变性,与适当引物杂交,再用DNA聚合酶延伸,克隆DNA。但该技术最终则是由美国Cetus公司人类遗传研究室的年轻科学家Kary B.Mullis于1985年发明,这一技术在很短的时间里即风行全球,形成了分子生物学领域的热潮,诸多科学家期望凭借这一工具来提高研究水平,解决所面临的一些难题。Mullis因其杰出的贡献,1993年获诺贝尔化学奖。
Mullis最初建立的PCR方法使用三种温度的水浴进行实验,用大肠杆菌DNA聚合酶I的KLENOW片段催化复性引物的延伸,但该酶不能耐受高温,所以每一轮反应都需要添加新的酶,产量不高且操作繁复,对实验操作要求较高,无法推广使用。
1988年Saiki等将耐热DNA聚合酶(Taq)引入了PCR技术。该技术才得到真正的应用。Saiki等利用PCR方法扩增人珠蛋白DNA,并用于镰刀状红细胞贫血的产前诊断。现在PCR技术以其高特异性、高敏感性及简便快捷使其成为基因诊断首选的技术之一。但PCR技术用于诊断也存在着明显的缺陷,理论上能将量极微的(fg DNA)目的基因在较短的时间内(1~2h)扩增到极易检测的微克水平。PCR经30~35个循环,扩增倍数一般可达百万倍。因此,PCR技术很容易出现污染而导致假阳性结果。
PCR技术问世以来正以惊人的速度发展,许多新型的PCR技术或由PCR衍生的新技术正不断出现,如转录依赖的扩增系统(TAS)、循环探针反应、链替代扩增(SDA)等。
2.技术要点
(1)PCR各组分的配制与加样量:
①引物浓度与引物的配制:
引物浓度:一般为10pmol,浓度太高会导致非特异扩增片段增多,浓度太低则扩增效率降低。
引物配制:厂家提供的引物一般是干粉状态并标明OD值,1 OD约含33μg。一般配置时稀释至10pmol/μl。
Oligo DNA中的每个脱氧核苷酸碱基的平均分子量近似为324.5U,则一条Oligo DNA的分子量=碱基数×324.5U。
例:您得到一管标为5 OD260的20 mer Oligo DNA
分子量=20×324.5 =6490U
质量数=5×33 =165μg
摩尔数=165/6490 =0.025μmol =25nmol
②模板DNA:浓度不可过高,20ng。
③Taq DNA聚合酶:一般用量1U/20μl反应体系。
(2)扩增轮数与退火温度
扩增轮数:一般30~40轮,30轮就可使模板扩增106倍。
退火温度快速计算公式:Tm值= (GC×4+AT×2)-5
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










