



实验五 λDNA的制备
用于亚克隆DNA的λ衍生载体主要是黏性质粒或丝状噬菌体载体。在这里,前两种方法描述了从中等及大规模的液体裂解物分离出噬菌体DNA,然后用密度梯度离心或者离子交换色谱来纯化噬菌体颗粒。第三种方法是分离噬菌体DNA的快速方法,适用于小规模液体裂物。
Ⅰ 密度梯度离心法和平衡梯度离心法
一、实验目的
学习从λ噬菌体裂解液制备λDNA的原理,掌握λDNA的制备方法。
二、实验原理
大规模的液体裂解用于制备大量的高纯度噬菌体DNA。噬菌体先通过氯化钅色梯度离心,然后平衡梯度离心从细胞残骸中分离。最后,有两种提取λDNA的方法供选择:在第一种方法当中,氯化铯靠透析去掉,然后用酚和氯仿抽提DNA;而第二种方法中,可用高等级甲酰胺直接从氯化铯纯化的噬菌体颗粒中提取λDNA。
三、实验仪器、材料与主要试剂
1.仪器
Beckman JA-10,JA-20,SW-28及Vti 50转头及离心瓶(管)、3mL注射器、带25-G针头、Beckman VTi 50快速封闭管。
2.材料
制备噬菌体裂解物、λ噬菌体滴定及DNA定量的材料。
3.试剂
5×聚乙二醇溶液、悬浮培养基(SM)、氯化钾、氯化铯溶液、氯仿、低盐缓冲液(0.05mol/ L NaCl,0.05mol/L Tris-HCl,pH7.5,0.01mmol/L MgSO4)、TM缓冲液(50mmol/L Tris-HCl,pH7.5,10mmol/L MgSO4)、缓冲酚(buffered phenol)、2mol/L Tris-HCl(pH8.5)/0.2mol/L EDTA(选择,供甲酰胺提取用)、甲酰胺(非常高的纯度,最好重结晶)、TE缓冲液(10mmol/L Tris-HCl,pH8.0,1mmol/L EDTA);
5.8g NaCl,2g MgSO4·7H2O,50mL 1mol/L Tris-HCl,pH7.5,0.01%明胶,加水至lL,高压灭菌;
CsCl溶液:
d=1.3g/mL:31.24g CsCl+68.76mL H2O
d=1.5g/mL:45.41g CsCl+54.59mL H2O
d=1.7g/mL:56.24g CsCl+43.76mL H2O
制备CsCl溶液的公式:%WW=137.48~138.11/d。这种计算方法假定CsCl中没有水。一般存放过长的试剂会从空气中吸收水分,这些贮存液的密度将比计算的要低些。然而,按我们的经验,噬菌体带总出现在梯度中间层。
四、操作步骤
1.制备和浓缩噬菌体
(1)用25mL液体裂解物制作1000mL裂解物。
(2)裂解物在JA-l0离心管中,以10000r/min(17700×g),4oC离心10min,去除细胞碎片。
(3)将上清液移至1000mL量筒中,加入5×PEG溶液至终浓度为1×。轻轻翻转混合,置4oC过夜。PEG溶液引起噬菌体沉淀。
(4)移出约50mL的上清液,并保留以备下步用。弃去剩余上清液,小心不要丢掉任何白色沉淀。
(5)将沉淀移至Nalgene离心管。用保留的上清液洗涤量筒,并加到离心管中于4oC,JA-20转头5000r/min(3000×g)离心10min。
(6)冰浴。去掉上层,小心不要丢掉任何白颜色的黏稠相,其中含有PEG和噬菌体。
(7)以最小体积的SM重新悬浮白颜色的黏稠相。转入一个125mL的锥形烧瓶中。加入悬浮培养基的体积不应超过白颜色的黏稠相的3倍。
(8)配制1mol/L KCl溶液。按相同的量,分4次加入,每次加入后混匀,冰浴l5~30min。加入KCl可慢慢沉淀PEG溶液,而噬菌体不沉淀。
(9)转移至Nalgene离心管中,在4oC,JA-20转头10000r/min(12100×g)条件下离心10min。PEG溶液会被沉降下来,而噬菌体留在上清液中。
(10)测定噬菌体滴度,并将上清液保存于16mm或18mm的玻璃试管中。噬菌体滴度应有1×1012~5×1013pfu/mL。分离噬菌体颗粒。
(11)将氯化铯梯度按以下步骤在一个SW-28离心管中灌注(图2-1(a)):
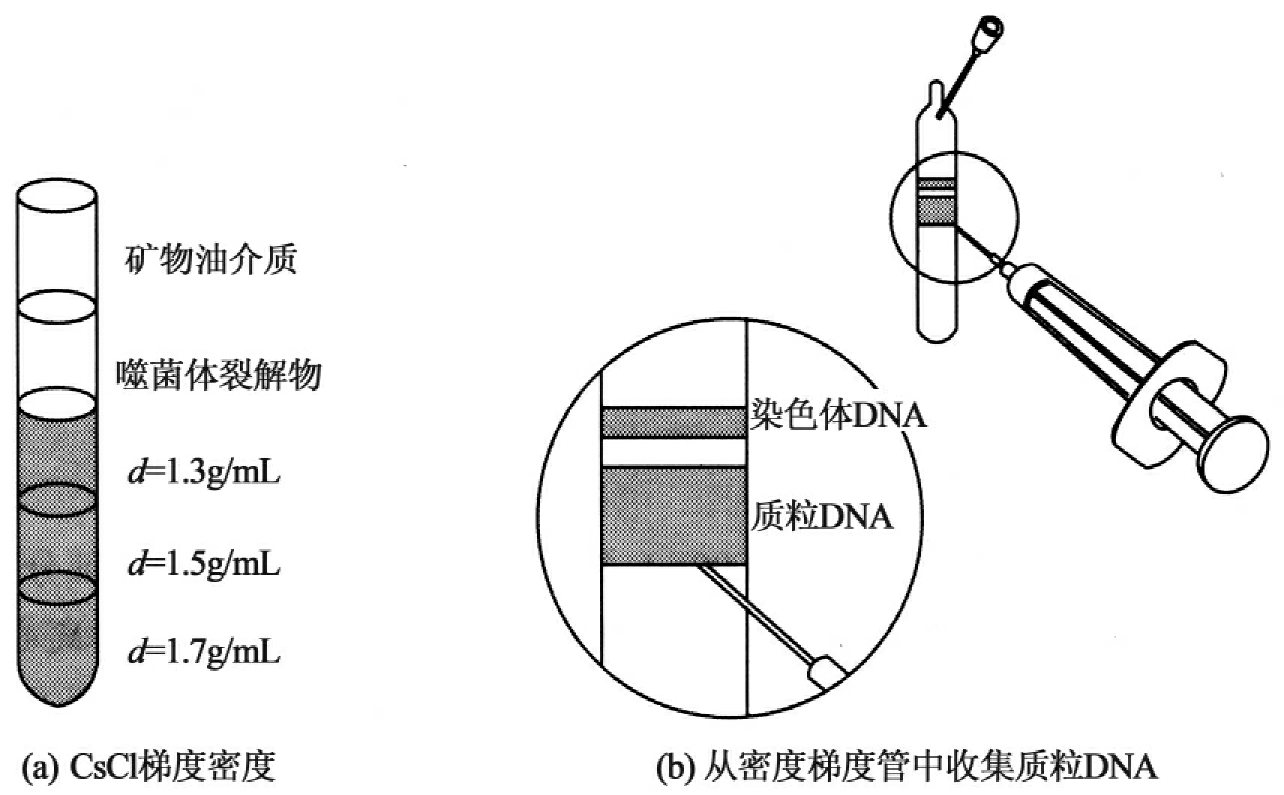
图2-1
第一层:3.5mL CsCl溶液,d=1.7g/mL
第二层:2.5mL CsCl溶液,d=1.5g/mL
第三层:2.5mL CsCl溶液,d=1.3g/mL
各层必须慢慢加入,避免混合。
(12)小心铺上λ裂解液[步骤(10)的上清液]在顶层。
(13)剩余的空间用SM装满,在4oC,104000×g(SW-28转头24000r/min)离心2h。
(14)用3mL注射器,25-G针头刺入试管条带下方,将其抽入注射器中[见CsCl/EB质粒制备图2-1(b)]。
通常可见3条带:一条是噬菌体带,蓝色,位于梯度最底处;一条是噬菌体外壳带,蓝色;以及一条白色细胞碎片带。偶尔有1~2条带都看不见。只要可见的带在d=1.5g/mL层,就没有问题。如果完全没有带,可能是噬菌体过少,没有必要继续做下去。如果层面不能清晰地区别,但蓝色带出现在大致正确的位置,回收带并称量裂解物的重量以测定密度。如果其密度约为1.5,那么可能是噬菌体。
(15)将噬菌体转入Beckman VTi50快速封闭试管中,用CsCl溶液注满试管(d=1.5g/ mL)。噬菌体颗粒重量大约有一半是蛋白质,另一半是DNA。噬菌体DNA比其蛋白质密度大些。因此,带有比野生型基因组大的噬菌体密度大于野生型基因组等长度的噬菌体,基因组小的噬菌体密度小些。
(16)4℃下81500×g(VTi50转头30000r/min)离心24h。
(17)用一支3mL带25-G针头的注射器,像在CsCl/EB质粒制备图2-1(b)所示那样,取出条带。应该仅有一条带可见。
噬菌体DNA萃取:以下提供从纯化噬菌体提取DNA的两个方法。在步骤(18a)~(21a),在酚和氯仿抽提DNA前,用透析方法去除氯化铯。在步骤(18b)~(21b),DNA直接从分离的噬菌体中用高级别的甲酰胺抽提。后者较快并柔和些。透析以及酚/氯仿抽提。
(18a)透析,搅动,于4℃500mL低盐缓冲液里至少4h,重复2次。
(19a)用等体积缓冲酚轻轻振荡20min,抽提3次。
(20a)用等体积氯仿萃取2次。
(21a)透析、搅动,置4℃500mL的TE pH8.0缓冲液中8h。换一次液,进行到步骤22。残存酚和氯仿用透析去除,不用乙醇沉淀,因为纯化噬菌体DNA重新悬浮很困难。
2.甲酰胺抽提
(18b)量出从步骤17得到的噬菌体条带的体积。加0.1倍体积2mmol/L Tris-HCl pH8.5/0.2mmol/L EDTA,颠倒混合。
(19b)加入1倍体积甲酰胺,混合,置室温下30min。
(20b)加入2倍体积的常温下的无水乙醇[每1倍体积相当于在步骤(18b)中噬菌体条带的原始体积],轻轻混匀,并离心1~2min。
(21b)弃去上清液并用70%乙醇洗涤沉淀。离心,再去除上清液,试管倒置片刻,用吸水纸吸干所有管壁上的乙醇液滴,用pH8.0的TE缓冲液溶解湿的沉淀块。
(22)测量DNA的浓度及纯度。
五、实验结果
测量DNA的浓度及纯度。常用分光光度法测定DNA和RNA的含量,具体操作如下:做DNA(或RNA)定量时,应在260nm和280nm两个波长下读数。在260nm的读数用于计算样品中的核酸浓度,OD值1相当于大约50μg/mL双链DNA、40μg/mL单链DNA或RNA及大约20μg/mL的单链寡核苷酸。可根据260nm以及在280nm的读数间的比值(OD260/ OD280)估计核酸的纯度。DNA及RNA纯品的OD260/OD280值分别为1.8和2.0,如果样品中污染有蛋白质或酚,其OD260/OD280将明显低于此值,此时无法对样品中核酸进行精确定量。
Ⅱ DEAE纤维素柱层析法
一、实验目的
学习从λ噬菌体裂解液制备λDNA的原理,掌握λDNA的制备方法。
二、实验原理
离子交换树脂优先结合粗制噬菌体裂解液中的污染物(大肠杆菌DNA、RNA及蛋白质),而噬菌体颗粒穿过交换柱而被分离,得到高纯度的噬菌体。然后用有机溶剂从纯化噬菌体中萃取DNA,并用乙醇沉淀。方法简单、快速,不需要DNase或CsCl密度梯度离心。获得高品质λDNA适于克隆、序列分析、限制酶切、连接以及体外包装。
三、实验仪器、材料与主要试剂
1.仪器
Beckman JA-14和JA-20转头及(或相当)15mL和13mL corex离心管、处理后的10mL注射器(1.4cm内径)或经过处理的1.5×10cm标准玻璃柱(Bio-Rad)玻璃纤维滤器或玻璃棉,另外还有用于液体裂解物或平板裂解物、λ噬菌体滴度、琼脂糖电泳以及酚抽提/乙醇沉淀的试剂和仪器。
2.材料
3.试剂
TM缓冲液(50mmol/L Tris-HCl pH7.5,10mmol/L MgSO4)、2%十二烷基磺酸钠(SDS选择)、0.1mol/L EDTA(选择)、氯化钠、聚乙二醇(PEG)8000、0.05mol/L HCl、10mol/L NaOH、DEAE纤维素(微粒阳粒子交换剂;Whatman ED5244057-050)、叠氮钠(NaN3)、5mol/ L NaCl、冰冷的纯异丙醇、TE缓冲液(10mmol/L Tris-HCl,pH8.0,lmmol/L EDTA)、25∶24∶1酚/氯仿/异戊醇、3mol/L乙酸钠,pH6.0、70%乙醇及冰冷的无水乙醇。
四、实验步骤
1.噬菌体粗裂解物的浓缩
(1)制备液体裂解物或平板裂解物及测定噬菌体滴度。裂解物应含1×1010~2×1010pfu/mL,总体积为200mL。当用平板裂解物时,从6个大的(150mm)或15个小的(10mm)培养皿中收集噬菌体于TM缓冲液(替代SM)中,并调整体积至200mL。另外,可用0.7%的琼脂糖凝胶检验在噬菌体裂解物中的λDNA。用2mL 2%SDS,2μL 0.1mol/L EDTA与20μL裂解液在室温下作用5min。凝胶电泳,包括分子量标准和对照(未处理)泳道。在该胶中,SDS/EDTA作用过的裂解物样品应当比未处理的样品显示出更为清楚的λDNA带。
(2)加入5.8g NaCl(终浓度为0.5mol/L)和20g PEG 8000(终浓度10%m/V)至200mL噬菌体裂解液中。如果噬菌体滴度低,有必要先加入NaCl使附着在碎片上的噬菌体释放。至4℃,5500×g(JA-140600r/min)离心10min去除碎片,然后加PEG。轻轻搅动PEG溶液,置于冰水中1h,这个时间能使大部分噬菌体沉淀。
(3)4℃,5500×g(JA-14,6000r/min)离心10min分离噬菌体沉淀(翻转瓶子,倒掉液体,并晃动以除残留的液体)。
(4)用3mL TM缓冲液重新悬浮噬菌体沉淀,并转移至一个15mL的corex离心管中,加30mL氯仿,轻轻混合,于4℃,3800×g(JA-20,5000r/min)条件下离心10min,收集上层含噬菌体的水相,不要搅动PEG界面。透过corex管,很容易观察PEG界面及分离上清液。
(5)加入3mL TM缓冲液至管中,混合并离心,如步骤4。保留水相部分并与上步得到的水相混合,用TM缓冲液调整体积至6mL。这一步的目的在于分离出PEG界面上的噬菌体颗粒。
2.DEAE纤维素柱的制备
(6)加入若干体积的0.05mol/L HCl以制备DEAE纤维素。确保溶液的pH值低于4.5。制备30~40根柱子所用足够的EDAE纤维素是很方便的,每根柱子需要约9mL的树脂(分离200mL噬菌体裂解液)。
(7)一边搅拌,一边加入10mol/L NaOH,直到pH值接近7.5。
(8)让树脂沉淀,吸出液体。用TM缓冲液平衡树脂(见说明)。
确保DEAE纤维素用TM缓冲液完全平衡。这可能需要重复该步骤4~5遍,此过程也去除了树脂中的细小颗粒。
(9)调整DEAE纤维素为75%树脂,25%TM缓冲液。加入NaN3至终浓度为0.02%,并置于4℃,可长期保存(数月)。
(10)取9mL树脂加到一支10mL的处理过的注射器(内径1.4cm×10cm或1.5cm×10cm)柱中,该柱子柱床高5~6cm,可分离200mL噬菌体裂解液。当采用注射器时,在底部放入一个玻璃纤维滤器或少许玻璃棉来支撑树脂。接上一段带有开关的小橡皮管于注射器头上,以此来控制液体流速。用标准玻璃柱,或市售柱来制备层析柱较为容易。
3.DNA分离
(11)在DEAE纤维素柱上加入步骤5所得的粗制噬菌体溶液6mL,并用10mL TM缓冲液洗脱。弃去最先的3mL洗脱液(外水体积),收集接下来的13mL于30mL的corex管中。13mL洗脱液也可按1mL来收集。一般噬菌体紧接着外水体积洗脱。由于散射作用,高滴度噬菌体的部分呈蓝色,这意味着柱子的分离效能很好。每部分取5μL进行琼脂糖凝胶电泳分析,即可判断噬菌体的纯度。
(12)加入2mL 5mol/L NaCl(400mmol/L终浓度)及10mL冰的纯异丙醇(40%终浓度)至13mL洗脱液中,置-20℃15min。
(13)4℃,3800×g(JA-14,5000r/min)离心10min,弃上清液。倒扣离心管直到液体完全流尽为止,用纸巾吸干离心管壁,不要碰到噬菌体沉淀。沉淀转移至1.5mL的微量离心管中。
(14)用酚抽提一次,酚/氯仿/异戊醇抽提两次,最后氯仿抽提。重复抽提,直至在界面没有可见的蛋白沉淀为止。
(15)加入0.1倍体积的3mol/L乙酸钠,pH6.0及2倍体积冰冷的无水乙醇,沉淀λDNA。一般来说,在室温下加入冰冷的乙醇后,λDNA立即形成丝状沉淀,白色的云状沉淀意味着留有蛋白质。λDNA可用一玻璃棒缠卷上,或通过轻轻搅拌浓缩成一球状颗粒。
(16)用70%乙醇洗涤DNA沉淀,吹干,重新悬于pH8.0的TE缓冲液中。待完全溶解后,测定DNA浓度,用TE调整至200μg/mL,储存于4℃。λDNA溶解可能要花费数小时。
五、实验结果
测量DNA的浓度及纯度。
Ⅲ 小规模液体溶解物制备法
一、实验目的
学习从λ噬菌体裂解液制备λDNA的原理,掌握λDNA的制备方法。
二、实验原理
该方法用于制备小量DNA。该方法制得的DNA可用于限制酶切分析。噬菌体通过离心浓缩,并用酚破坏其包被,然后用乙醇沉淀DNA。
三、实验仪器、材料与主要试剂
1.仪器
离心机、旋涡振荡器。
2.材料
噬菌体裂解产物。
3.试剂
5mg/mL DNase、10mg/mL不含DNase的RNase、0.05mol/L Tris-HCl,pH8.0、3mol/L乙酸钠,乙酸调节pH值到4.8。
四、实验步骤
1.约50mL噬菌体裂解液中,加入10μL 5mg/mL DNase和25μL 10mg/mL无DNase的RNase,37℃孵育1h。降解在裂解时释放的DNA和RNA,混合物的黏度将降低。
2.在4℃,132000×g(SW-28转头27000r/min)条件下离心1.5h。另外,可在,48000×g(JA-20转头20000r/min)条件下离心2.25h沉降噬菌体。这种方式获得的沉淀更容易重新悬浮。
3.弃去上清液,倒扣试管于一吸水纸表面上,用纸巾吸去管壁上的水,去除任何残留液体,重新悬浮噬菌体沉淀于200μL 0.05mol/L Tris-HCl,pH8.0中(将试管翻转后,可看见小块的透明沉淀)。
4.将溶液转移至一微量离心管中,加入200μL缓冲酚,旋涡振荡20min,或在微量离心管摇动器中摇20min,离心2min,保留水层(上层),重复酚抽提。酚能使噬菌体包膜变性,并释放DNA,这些包膜蛋白在酚/水界面上形成很厚的白色沉淀。一定要剧烈搅动,沉淀不会再悬浮。在第二次酚抽提后,应只有较少的白色沉淀。如果在界面上仍有大量的沉淀,再抽提。
5.加200μL氯仿,摇匀,离心,保留水相(上相),重复。
6.加20μL 3mol/L乙酸钠,2倍体积无水乙醇于室温沉淀DNA,12000r/min离心10min。
7.去除上清液,加1mL70%乙醇洗涤沉淀,12000r/min离心5min。
8.去上清液,真空干燥,100μL TE缓冲液,溶解DNA。如果沉淀仍然湿润,DNA很快就会被溶解,可用3μL DNA溶液做限制酶切。
五、实验结果
测量DNA的浓度及纯度。
六、思考题
1.比较λDNA制备的不同方法。
2.如果用平板裂解法制备λDNA,上层固体应该用琼脂糖还是用琼脂?
七、方法评注
1.背景知识
以上提供了3种从完整的噬菌体颗粒中抽提和纯化DNA的方法。3种方法在使用粗裂解物的量、噬菌体在裂解释放DNA前的浓缩、纯化方式是不同的。例如,文库构建需要大规模制备大量高纯度的DNA(超过200μg)。这种方法仍基于分离λ噬菌体的传统方法:用聚乙二醇(PEG)从裂解液中沉淀噬菌体,并随之用CsCl梯度及平衡密度梯度离心纯化。在大量的裂解物中,存在相当数量的细胞碎片,用两种不同的梯度来进行纯化是有必要的。接下来,有两种从噬菌体包膜中提取DNA的方法可供选择:其一,噬菌体蛋白质通过一系列酚抽提去除,最后噬菌体DNA在TE缓冲液中透析以去除任何残留的酚或氯仿;其二,用高等级甲酰胺将DNA直接从包膜中萃取出来。
另外,还有一种中等规模的制备方法。这种方法得到的纯λDNA(约200μg)足够用于大多数标准DNA操作,如克隆、测序、体外包装及分子杂交。用离子交换色谱将噬菌体颗粒从裂解物的细胞成分中分离出来,一步就可以得到高度的纯品。色谱纯化工作原理如下: DEAR(二乙氨乙基)纤维素是一种阴离子交换树脂,它有阳离子活性基团,能在接近中性pH值和中等的离子强度的缓冲液中吸附负电荷基团,在噬菌体裂解物中主要的细胞污染物属于负电荷分子(DNA、RNA和蛋白质),它们优先被吸收到DEAR-纤维素的正电荷基团上。
曾报道过用Whatman DE3阴离子交换树脂纯化噬菌体的两种不同方法。在这里所描述的方法中,细胞污染成分结合到离子交换树脂柱上,而噬菌体颗粒不结合。由于核酸的抽提没有使用SDS及蛋白酶K,所得的DNA勿需进一步纯化即可用于限制性内切核酸酶及DNA修饰酶的作用,并曾被成功地用于制备基因组表达文库。另外,也可以将噬菌体颗粒与树脂结合,然后用高盐缓冲液洗脱,这种方法用于同时处理几个克隆,以获得数微克的DNA来进行亚克隆。
快速、小规模的噬菌体纯化方法可得到小量中等纯的DNA。这种DNA适于限制酶酶切分析。该方法用高速离心沉降噬菌体,RNase和DNase破坏宿主核酸(这些酶对噬菌体DNA没有作用,因为它们还包装在噬菌体头部)。接下来,噬菌体DNA用酚从包膜中抽提出来,乙醇沉淀浓缩。
2.重要参数
无论任何方法,分离λDNA失败的原因大多是因为在起始的裂解物中噬菌体滴度低。因此,首先滴定裂解物可以避免浪费时间。可用平板稀释法滴定噬菌体,如果滴定少于109pfu/mL,重新制作裂解物。另一种常用来检查DNA量的方法是取20μL裂解物,经SDS/ EDTA处理后,在琼脂糖凝胶上电泳。凝胶电泳方法不费时,不论是否见得到噬菌体颗粒,都能较准确地估计DNA量。图2-2(泳道3)显示的是一典型的凝胶样本,从上面可看出存在于裂解物中的细胞核酸及λDNA(约50ng)的相对量,肉眼可以看到≥10ng的DNA条带,建议使用新鲜的裂解物。
在层析纯化中,裂解物与柱子大小的比率是一个重要变量。一个9mL的柱床体积足够用于200mL的裂解液(1010pfu/mL水平);如果裂解液过多(或者如果裂解物的细胞碎片过浓),柱床体积应当相应提高,如柱子超负荷,细胞DNA及RNA可以连同噬菌体一并洗脱(欲要检查是否过载,用琼脂糖电泳监测粗裂解液的过柱滤液)。如果DNA被污染了,将滤液集中在一起,在有机溶剂抽提之前,用DNase和RNase处理。应按照Whatman的产品说明,小心制备、平衡及包装DEAE纤维素,注意DEAE纤维素型号(Whatmann ED52),以及pH值和缓冲液离子强度(用提供的TM缓冲液配方),这很重要。氯仿裂解培养物常含有很黏稠的相对分子质量高的染色质DNA,有可能堵塞DEAE柱。这样的裂解物可以在上样前用DNase处理。
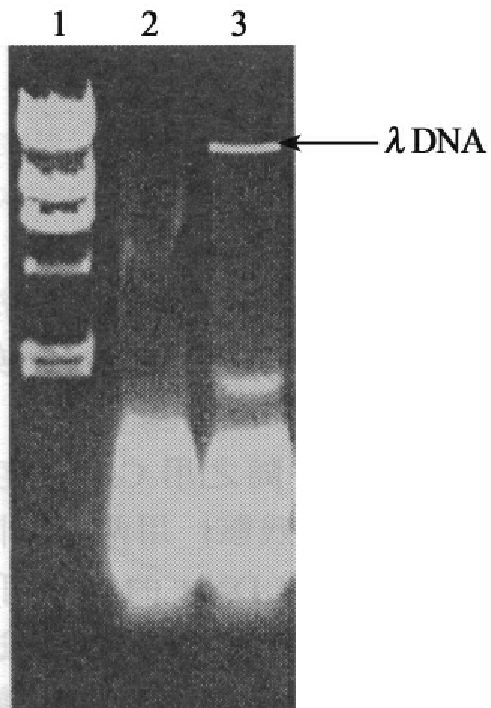
图2-2 λ噬菌体裂解物的琼脂糖微型凝胶电泳
1λDNA用HindⅢ消化
2未经SDS和EDTA处理的20μL裂解物
3用2%SDS和0.1mol/L EDTA处理的20μL裂解物
在有机萃取后,先透析,再沉淀噬菌体DNA,因为大分子的DNA很难溶解。在被分离出来后,λDNA因相对分子质量大而容易被剪切。如需要完整的DNA(例如,当噬菌体臂要用来建文库时),应当特别小心地处理DNA,非常轻柔地摇动、搅动或旋涡振荡以混合。
3.预期结果
每种方法得到的λDNA取决于噬菌体滴度和粗裂解物体积。如果起始裂解物滴度为1×1010~2×1010pfu/mL,那么1mL裂解物应产出约1μg DNA,也就是说,2×1010个噬菌体颗粒得到约1μg纯噬菌体DNA。因此,大规模的制备方法,其所用裂解液多是1000mL,那么就应得到约1mg的噬菌体DNA,DEAE方法(200mL裂解液)约可得到200μg DNA,小规模法(50mL裂解物)约可得到50μg DNA。
4.时间安排
大规模制备到梯度纯化,一般至少要3d时间:第1天约30min用来PEG沉淀;第2天约5h的离心沉降和收集沉淀、梯度及平衡梯度离心;第3天约30min用以收集和酚抽提噬菌体带,然后持续8h的2个透析步骤。噬菌体裂解液可作为中间产物储存于4℃数月而不至滴度下降。小规模制备花费约4~6h,可一次处理多个样品。
从液体裂解物或平板裂解物开始,柱纯化噬菌体及DNA分离可在4h内完成,上柱的噬菌体粗制液的制备花费约2h。装柱、上样、洗脱及DNA抽提还需要2h,通常可以同时处理4~5个不同的柱子而毫无问题。DEAE纤维素可预先处理好,储存于4℃数月。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










