



Ⅰ 苯乙酮的合成
反应式
![]()
实验步骤
1.常量合成
在100mL三口烧瓶上,装上回流冷凝器,在冷凝器的上口接一个装有无水CaCl2的干燥管并连接气体吸收装置,在烧杯中加入5%NaOH溶液作为吸收剂,吸收反应中产生的HCl气体。出气口与液面距离l~2mm为宜,千万不要全部插入液体中,以防倒吸。在三口烧瓶的另一个口上装上恒压滴液漏斗,装好电磁搅拌器。向反应瓶中加入12g无水三氯化铝和16mL苯,开动搅拌器,边搅拌边滴加4mL (0.042mol)乙酸酐,开始先少加几滴,待反应发生后再继续滴加。此反应为放热反应,应注意控制滴加速度,切勿使反应过于激烈,必要时可用冷水冷却。此过程需10~15min,待反应缓和后,用水浴加热反应瓶并搅拌,直至无HCl气体逸出为止。此过程约需40min。
将反应瓶置于冷水浴中,边搅拌边慢慢滴加18mL浓盐酸和40g碎冰。若还有固体存在,应补加浓盐酸使其溶解。然后将反应液倒入分液漏斗中,分出上层有机相,用40mL石油醚分2次萃取,萃取后的石油醚与有机相合并,依次用10mL 10% NaOH溶液和10mL水洗涤。在水浴上蒸出石油醚和苯后,再用常压蒸馏或减压蒸馏蒸出产品。
常压蒸馏时,当温度超过140℃时改用空气冷凝器继续蒸馏,收集198~202℃的馏分,产品为无色透明液体,产率约为65%。
2.半微量合成
在50mL的三口烧瓶上,装上回流冷凝器,在冷凝器的上口接一个装有无水CaCl2的干燥管并连接气体吸收装置,在烧杯中加入5%NaOH溶液作为吸收剂,吸收反应中产生的HCl气体。出口与液面距离1~2mm为宜,千万不要全部插入液体中,以防倒吸。在三口烧瓶的另一个口上装上恒压滴液漏斗,装好搅拌器。
向反应瓶中加入6g无水三氯化铝和8mL苯,开动搅拌器,边搅拌边滴加2mL乙酸酐,开始先少加几滴,待反应发生后再继续滴加。此反应为放热反应,应注意控制滴加速度,切勿使反应过于激烈,必要时可用冷水冷却,此过程约需10min左右。待反应缓和后,用水浴加热反应瓶并搅拌,直至无HCl气体逸出为止。待反应液冷却后进行水解,将反应液倾入盛有10mL浓盐酸和20g碎冰的烧杯中(此操作最好在通风橱中进行),若还有固体存在,应补加浓盐酸使其溶解。然后将反应液倒入分液漏斗中,分出上层有机相,用30mL石油醚分2次萃取下层水相,合并有机相,依次用5mL 10%NaOH溶液和5mL水洗至中性。用无水硫酸镁干燥。在水浴上蒸出石油醚和苯后,再用常压蒸馏或减压蒸馏蒸出产品,常压蒸馏收集198~202℃的馏分。产品为无色透明液体,产率约为65%。
纯苯乙酮沸点为202℃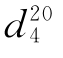 为1.0281,
为1.0281,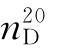 为1.5338,红外光谱如图5-22所示。
为1.5338,红外光谱如图5-22所示。
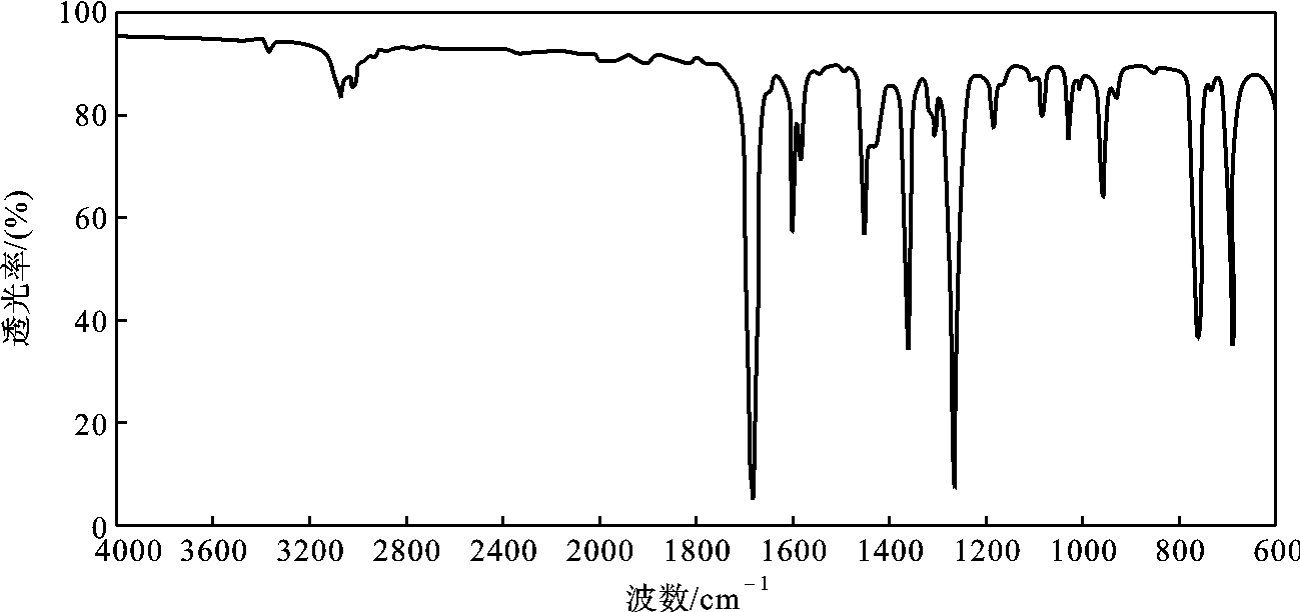
图5-22 苯乙酮的红外光谱图
注意事项
(1)此实验应在无水条件下进行,所用试剂及仪器需要严格干燥。无水三氯化铝在空气中容易吸潮分解,在称量过程中动作要快,称完后及时倒入烧瓶中,将烧瓶和药品瓶盖子及时盖好。苯用无水CaCl2干燥过夜后再用。放置时间较长的乙酸酐应蒸馏后再用,收集137~140℃之间的馏分。
(2)在与无水三氯化铝接触的过程中,应避免与皮肤接触,以免被灼伤。
(3)反应温度不宜过高,一般控制反应液温度在60℃左右为宜,反应时间长一些,可以提高产率。
(4)加乙酸酐时,开始应慢一些,过快会引起暴沸,反应高峰过后可以加快速度。
思考题
(1)为什么要用过量的苯和无水三氯化铝?
(2)为什么要用含酸的冰水来分解产物?
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。


![怎样释[蒸汽机]](https://file.guayunfan.com/2020/zb_users/upload/2020/09/9.jpg)








