



三、OV在肿瘤细胞内特异性增殖的机制
(一)选择性进入细胞
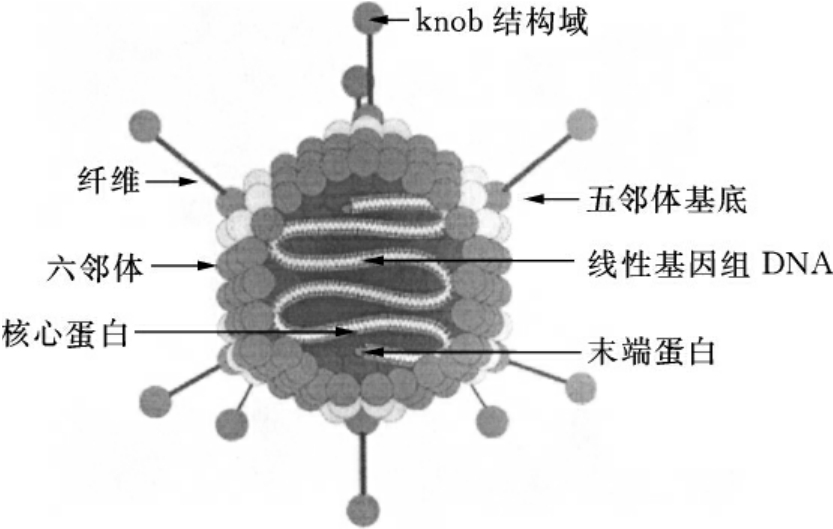
图16-7 腺病毒结构示意图
腺病毒为二十面体对称结构,直径为70~100nm。毒粒具有252个壳粒,顶角壳粒为12个五邻体(penton),一个基底(base),每一个纤维蛋白(fiber)坐落在一个五邻体上,此外还有240个非顶角壳粒,称为六邻体(hexon)。腺病毒感染靶细胞时,由病毒衣壳纤维蛋白中的knob结构域识别并结合细胞表面的受体(CAR),然后五邻体蛋白与细胞表面的整合素家族分子相互作用,内化进入细胞,因此CAR对于腺病毒的感染至关重要(图16-7)。但是,这种感染缺乏特异性,而且CAR在许多肿瘤细胞表面并不表达,导致腺病毒不能感染这些细胞。提高腺病毒感染的效率和靶向性,构建一类双特异性腺病毒导向分子,其一端为抗腺病毒纤维蛋白(如knob结构域),或五邻体蛋白的抗体,或CAR的胞外区;另一端为与肿瘤细胞表面受体结合的配体,或肿瘤特异性抗原的抗体,这种双特异性分子在腺病毒和肿瘤细胞间起连接作用并提高腺病毒感染的特异性。Haisma等将抗腺病毒纤维蛋白抗体与抗EpCAM抗体融合,构成双特异性抗体,EpCAM是特异表达于腺癌细胞表面的肿瘤抗原,因此这种双特异性抗体高效地介导腺病毒载体感染的原发的和转移至肝脏的结肠癌细胞,而不感染正常肝细胞。
腺病毒纤维蛋白由尾部(tail)、茎(shaft)及头部(knob)组成,亲和性由头部决定,头部由AB、CD、DE、FG、HI及IJ6个环组成,不同血清型的腺病毒,HI环长度变化最为明显。纤维蛋白位于腺病毒的外壳突起,与细胞受体直接结合。同时,纤维蛋白的改造不会影响腺病毒包装,因此实现腺病毒靶向感染的另一种方法是将腺病毒的纤维蛋白进行改造,在纤维蛋白的C-端或knob结构域的HI环处融合一段能结合肿瘤细胞的肽段。Dmitriev等将来源于血管内皮细胞的RGD-4C肽段(CDCRGDCFC),取代腺病毒纤维蛋白knob结构域HI环的部分氨基酸序列,表达这种嵌合衣壳蛋白的腺病毒对肿瘤细胞感染的特异性和效率都显著提高;而在AB、CD及DE环中突变,可使腺病毒对腺病毒受体(CAR)的亲和力下降千倍以上,使腺病毒不易被正常细胞CAR结合。此外,用Adv11或Adv35的纤维蛋白替换Adv5的纤维蛋白,可增加它对白血病细胞的导入性。
(二)肿瘤选择性复制
构建肿瘤内选择性复制的溶瘤病毒有几个不同的策略,主要是根据肿瘤细胞内异常的信号传导途径或者肿瘤细胞特异性启动子调控病毒复制进行设计。
1.OV靶向p53途径 正常细胞与肿瘤细胞存在着某些基因表达上的差异,某些病毒在正常细胞内增殖所必需的基因,在肿瘤细胞内并不需要,因此,去除这些基因后病毒在肿瘤细胞内仍可特异性复制,而在正常细胞内则不能复制,从而大量杀伤肿瘤细胞。
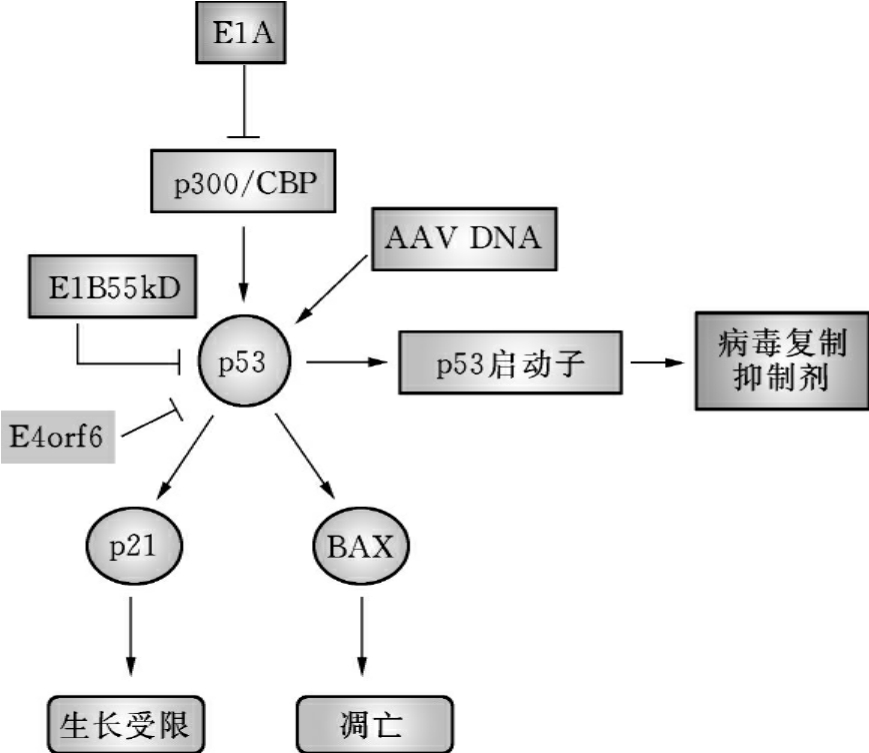
图16-8 OV靶向p53途径策略
(引自Antonio Chiocca,Nat Rev Cancer,2002)
正常细胞被腺病毒感染后并没有立刻出现凋亡,因野生型腺病毒存在抑制p53激活的蛋白,即E1B55×103(55kD)蛋白(图16-8),使野生型腺病毒能在正常细胞内增殖。缺失突变E1B55×103(55kD)基因的腺病毒感染细胞后,不能抑制p53的激活,使细胞很快凋亡,病毒不能再增殖,从而使感染终止;当此病毒感染p53突变或失活的肿瘤细胞后,不能促使细胞凋亡。但能在肿瘤细胞内大量复制及增殖,导致肿瘤细胞溶解死亡,并释放新的病毒感染其他肿瘤细胞。McCormick F等应用E1B 55×103(55kD)缺失的腺病毒ONYX-015对5株p53正常的细胞株进行实验,发现无明显的杀伤作用;而对p53功能异常的肿瘤细胞株具有很强的杀伤作用。E1B55×103(55kD)蛋白也与病毒晚期蛋白的翻译和运输有关,使病毒在肿瘤细胞内的复制能力和溶瘤效果下降。
除了病毒蛋白E1B55×103(55kD)可以抑制p53活性外,E1A病毒蛋白能抑制p53的共激活因子p300/CBP的活性,E4orf6能结合并抑制p53活性(图16-8)。因此改造病毒多个基因可以提高病毒的选择性感染。以腺病毒01/PEME为例,这个病毒中插入依赖p53表达的E2F抑制蛋白基因,选择性地削弱病毒在正常细胞中的复制能力。此外,E1A-CR1突变,会失去结合并抑制p300/CBP能力,为了增强病毒复制和对肿瘤杀伤能力,在E3区引入了病毒主要晚期启动子(MLP),使E311.6×103(11.6kD)过表达。实验证明,与ONYX-015相比,01/PEME在肿瘤细胞内的复制能力明显增强。
各种不同的病毒由于基因组特异的二、三级结构,在p53-肿瘤细胞中有选择性复制。AAV是单链DNA病毒,它感染的p53+正常细胞进入G2/M期停滞,而感染p53-肿瘤细胞将发生凋亡,病毒的这种作用类似于破坏DNA结构消灭肿瘤的化疗试剂的作用。此外,具有基因组特异结构(如发卡结构、高GC含量的单链DNA、双链RNA等)的其他病毒,对p53-的肿瘤细胞有不同程度的选择性。
2.OV靶向p16/RB途径 p16/RB途径调控细胞周期的起始及进程,破坏这个途径的一个或多个因子能导致细胞癌变。p16是这个信号途径的上游效应因子,是CDK4-CDK6-Cyclin D复合物的抑制剂。后者能增强RB磷酸化,超磷酸化的RB释放转录因子E2F,E2F则介导多种涉及G1/S进程的基因转录(图16-9)。p16抑制CDK4-CDK6-Cyclin D复合物的活性,使RB处于低磷酸化,防止RB磷酸化而激活E2F。在p16/RB有缺陷的肿瘤细胞中,E2F被激活,其靶向激活的基因编码许多与核酸代谢相关的蛋白,如二氢叶酸还原酶(DHFR)、胸苷激酶(TK)、核糖核苷酸还原酶(RR)和胸苷合成酶(TS),这些蛋白使细胞通过G1/S检测点,进入S期,便于病毒复制。
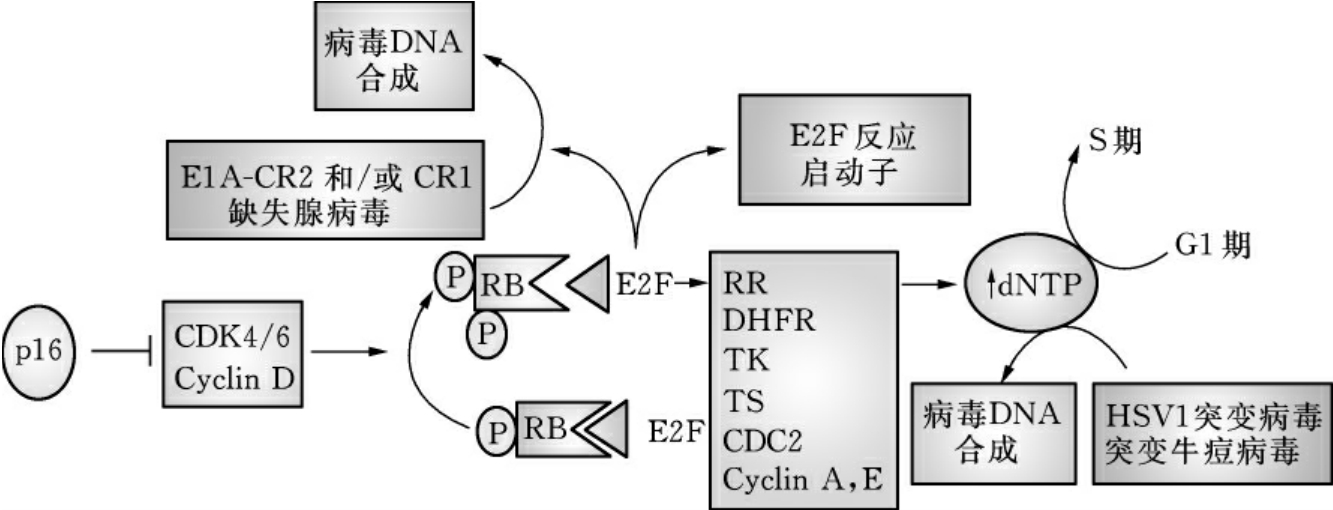
图16-9 OV靶向p16/RB肿瘤抑制蛋白途径
(引自Antonio Chiocca,Nat Rev Cancer,2002)
RB也能被病毒蛋白如腺病毒E1A、SV40的T-antigen和HPV的E7灭活,从而使细胞进入S期便于病毒更加有效地复制。以腺病毒为例,E1A-CR2删除的腺病毒突变体在RB缺失的肿瘤细胞内复制效率比生长停滞的正常细胞高100~1000倍。将腺病毒的E4、E1的启动子换成E2F启动子,同时删除E1A-CR2的腺病毒突变体,从3个水平来调控病毒的复制,这种OV无论在细胞实验还是动物实验都增强了肿瘤选择性杀伤效应。
HSV1和痘病毒表达的某些蛋白在细胞分裂状态下属于冗余蛋白,但这些蛋白使病毒在静息期细胞内也能复制。例如,HSV1的TK和RR在细胞处于静息状态时不表达,在细胞周期的G1/S期时被上调。痘病毒表达同HSV1相似功能的蛋白,使病毒在分裂后期的神经元细胞内复制。依据这一特点构建缺陷这些基因功能的HSV1或痘病毒突变体,例如,突变UL39编码的RR基因产生的HSV1突变体能在p16/Rb途径缺陷的肿瘤细胞内选择性复制,产生的病毒滴度比正常细胞内高100倍。这种基因缺陷的病毒是开发HSV1溶瘤病毒的良好开端。
另一种HSV1突变体Myb34.5,它的ICP34.5基因在B-Myb启动子控制下。B-Myb是E2F调控的转录因子,其启动子中含有E2F的结合位点。这种OV从两个水平依赖E2F的活性,一是ICP34.5肿瘤依赖性表达,一是弥补UL39的缺陷,它在肿瘤细胞内产生的病毒滴度比在正常细胞内高出1000~10000倍。在鼠转移性肝癌模型中,病毒经静脉给药,使肿瘤体积缩小,也使转移病灶消失,而且也没有明显的不良反应。
类似上述机制,删除TK基因的HSV1和痘病毒也有在肿瘤细胞选择性复制和溶瘤特性。因为哺乳动物的TK转录也受E2F调控,因此这种突变体也靶向p16/RB途径。Martuza等将HSV1复制必需的核糖核苷酸还原酶或胸腺嘧啶脱氧核苷激酶(HSV-tk)基因失活,这样病毒只能在上述酶含量丰富的、分裂迅速的癌细胞内增殖,而在正常中枢神经细胞内不能增殖。因此,该病毒能有效溶解胶质瘤细胞,而对正常细胞几乎没有作用。为确保安全,他们又将导致脑炎的病毒基因剔除,这种单纯疱疹病毒G207几乎对所有实体肿瘤均有明显的疗效。
总之,特异性靶向p16/RB途径的OV,都是针对病毒调控核酸代谢所必需的基因,这些基因突变有利于病毒在分裂的细胞内选择性复制;另一种将编码病毒必需蛋白的基因(腺病毒E1和E4,HSV1的ICP34.5)置于E2F启动子控制下,以利于病毒在RB途径缺陷的肿瘤细胞内特异性复制。
3.OV靶向RAS/PKR或IFN/PKR途径 双链RNA(dsRNA)依赖的蛋白激酶(PKR)和它相关的信号通路调控各种OV的肿瘤特异性复制能力。病毒感染细胞后不仅激活了p53应答,同时也激活另一个应激反应——PKR应答(图16-10)。PKR是胞内蛋白激酶,它能使翻译起始因子eiF2α磷酸化,从而关闭细胞内蛋白合成。PKR N端与dsRNA结合而激活,PKR的活性也可以通过各种细胞因子(如IFN)而不依赖dsRNA激活。因此抑制肿瘤细胞PKR活性是病毒选择性复制的关键。
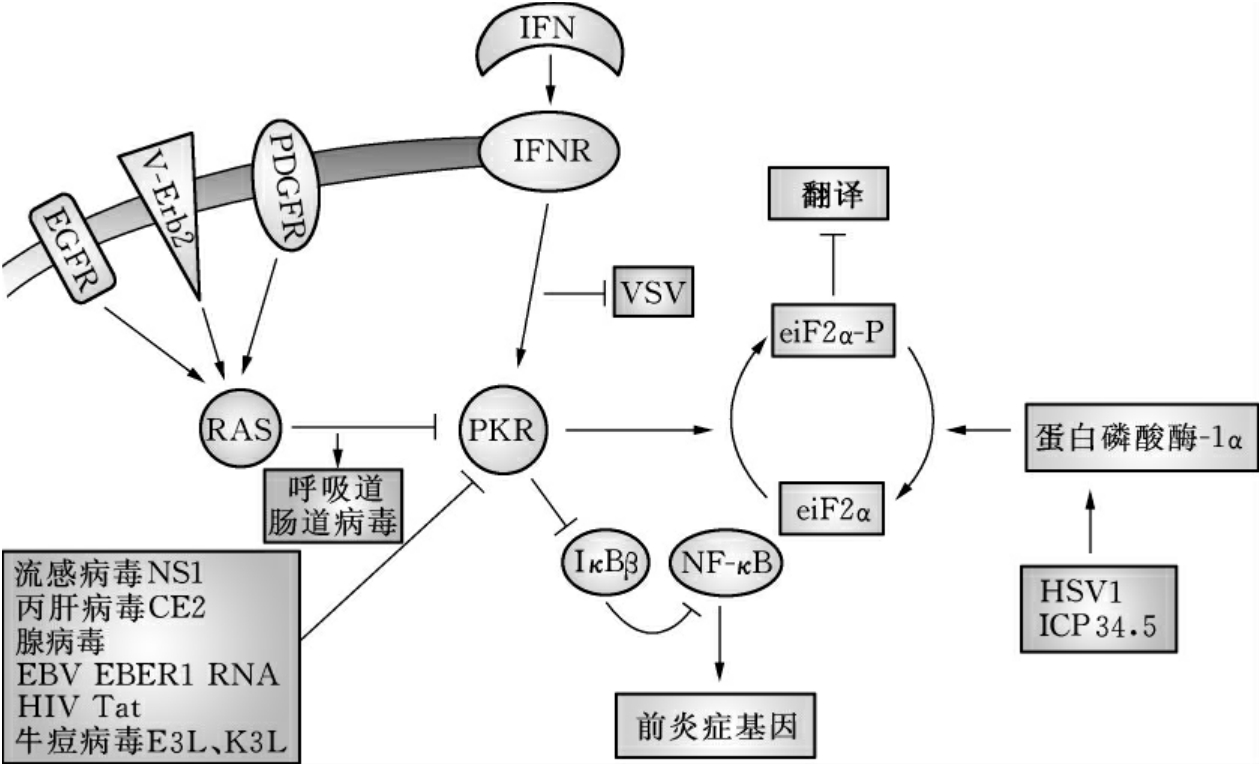
图16-10 OV靶向RAS/PKR或IFN/PKR途径
(引自Antonio Chiocca,Nat.Rev.Cancer,2002)
很多肿瘤细胞RAS处于激活状态,通过信号通路使PKR去磷酸化而抑制PKR的活性,eIF2α不能被磷酸化,则细胞内蛋白能够合成,野生型dsRNA病毒如呼肠孤病毒(reovirus)和新城疫病毒(newcastle disease virus)则在肿瘤细胞内增殖,从而特异地杀灭RAS高表达的肿瘤细胞。PKR的抑制蛋白(如HSV1的ICP34.5、流感病毒NS1等)基因突变的病毒能够在RAS过表达的肿瘤细胞内复制,而在正常细胞中PKR限制了这些突变体的复制。
IFN-α和-β是由各种细胞包括成纤维细胞,巨噬细胞和肿瘤细胞分泌产生的细胞因子,它们与受体结合后,通过下游的效应分子激活PKR。许多肿瘤的IFN/PKR信号通路有缺陷,病毒突变体就能在这些信号缺陷的细胞中选择性复制。呼吸道肠道过滤性病毒(reovirus)和水疱状口腔炎病毒(VSV)也能特异性地在IFN/PKR信号通路缺陷的肿瘤细胞内复制,是天然的溶瘤病毒。
4.肿瘤细胞或组织特异性启动子调控病毒复制 肿瘤细胞或组织特异性启动子或增强子中插入病毒增殖必需基因,控制病毒增殖必需基因的转录,使病毒在肿瘤细胞内特异性增殖。这类启动子主要有:①针对肿瘤特征的启动子,如缺氧诱导因子1(HIF-1)启动子、人的端粒酶(hTERT)启动子、细胞S期特异性启动子(如E2F启动子)、p53启动子、b-Myb启动子和TCF4启动子等;②针对个别肿瘤特征的启动子,如肝癌的甲胎蛋白(AFP)启动子、胃癌和结肠癌的癌胚抗原(CEA)启动子、乳腺癌或卵巢癌的DF3/MUC-1启动子、黑素瘤的酪氨酸酶(Tyrosinase)启动子、肺癌的SPB(表面活性剂蛋白B)启动子等;③针对某一类组织特异性启动子,如前列腺细胞特异性抗原(PSA)启动子,杀灭这类组织并不会危及生命,如前列腺细胞、卵巢细胞、某种类型的淋巴细胞等。
以编码甲胎蛋白基因的启动子为例,甲胎蛋白在胚胎发育早期组织中表达,成年组织局限于肝来源的肿瘤组织。AFP启动子控制E1A或E1B表达的溶瘤腺病毒,在AFP阳性的细胞内复制能力比AFP阴性的细胞增加10000倍。静脉给药治疗实验性小鼠肝癌能使肿瘤消退,而对正常组织没有严重的不良反应。肝硬化细胞也有很高的AFP,影响了AFP启动子的广泛应用。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










