



2 保障儿童用药安全有效的制度及规范研究[8]
一、引言
据第五次全国人口普查统计表明,我国14岁以下的儿童有2.9亿人。儿童的身体健康是关系到国家未来的大事,儿童的用药状况是影响儿童健康的一个重要因素。儿童处于生长发育期,生理特征独特,对药物具有特殊的反应性和敏感性,药动学、药效学与成人相比有很大的差异。
WHO提出理想的儿童药物应适合于不同年龄、生理条件和体重的服药儿童,并有灵活的固体口服剂型,可全部服用,用各种液体溶解,或撒在食物上,使儿童易于服用。但是,目前儿童用药现状不容乐观,存在诸多问题。许多基本药物没有儿童剂型,用于儿童的许多药物在疗效和安全性方面缺乏足够的儿童信息等现象普遍存在。同时,在我国相关法规、制度处于真空期,在有关保护儿童权益、促进儿童健康发展的两个十年发展纲要中,有关儿童健康的内容均未涉及儿童用药领域。而以美国为代表的发达国家通过立法逐步建立了较为完善的儿童用药管理制度,受到社会注重并已取得较好的社会、经济效益。探讨如何在我国逐步建立、完善相关制度、规范,保障儿童用药安全、有效,其意义重大。
二、我国儿童用药现状及问题
(一)研究方法—文献检索
以“药物不良反应”或“药品不良反应”和“儿童”;“政策”或“法规”或“制度”或“规范”和“儿童用药”或“儿科用药”;“药品说明书”和“儿童”或“儿科用药”等为关键词检索历史文献,检索、分析历史文献,对目前我国儿童用药情况进行总体梳理。
(二)文献检索的结果
1.品种与规格
目前国内市场90%的药物都没有儿童剂型,而患病儿童占总患病人数的20%左右[1]。《中国新药实用全集》中新药品种共2 275种(含中药),儿童用药专用品种仅40种[2]。由于儿科制剂批量小、成本高、利润低,故生产儿童用药的专用厂家寥寥无几,临床上不得不将成人剂型、规格的药品分割后用于儿童,导致剂型破坏、剂量不准确,对于治疗指数低、安全范围窄的药物,危险性更大。20世纪90年代初上海和杭州医药公司供应药品目录中,儿童规格占9%左右。上海某医院儿科药房2005年8月~2006年8月所使用的141个口服制剂中,适宜儿童口服的药品41个(29.08%),儿童专用药品(说明书注明主治儿童疾病或仅有儿童用法的药品)34个,占24.11%,同时期使用的69个注射剂中仅小儿氨基比林为儿童专用药品。药品最小规格剂量单位再分零现象普遍存在。上海某儿童专科医院2005年心胸外科住院患儿出院带药中卡托普利≤1/4量的处方数占到总数的59.76%,还有部分处方的分零情况达到1/10量甚至1/20量[3]。浙江大学医学院附属儿童医院,90年代初618种库存药品中儿童药品只有36种(5.8%),2006年该院库存药品1 103种,儿童药品也有所增加,达到64种,但百分比仍为5.8%[4]。武汉市妇女儿童医疗保健中心,2008年库存的738种药品中,专门针对儿童设计的剂型与规格药品为65种,占8.8%[5]。儿童用药品种少,专用剂型、规格少的现象目前是很严重的,已引起临床医生、药师的重视。但是虽然儿童药品有所开发,儿童口服制剂虽有所增加,但针剂和注射剂中几乎没有儿童剂型[6]。
2.药物不良反应
儿科中导致ADR的常见药物中抗感染药总是占首位(45.73%[7]、45.92%[8]、52.0%[9]、63.92%[10]、83.75%[11]),抗感染药物中头孢菌素类药物ADR发生率居前(38.81%[11]、42.1%[12]、46.67%[8]);其次多为中药制剂(9.18%[8]、10.55%[7]、16.2%[12]、22.68%[10]),中枢神经系统用药(20.8%[9])。静脉用药是引起ADR的主要给药途径(49.9%[9]、53.5%[7]、62.24%[8]、72.1%[12]、92.5%[11]、96.91%[10]),合并用药引起ADR(80%左右)高于单一用药(20%左右)[8,12]。ADR好发于学龄前儿童(59.2%[8]、70.6%[7]、75%[11,12]),以过敏反应为多,多累及皮肤及其附件(43.97%[7]、55.1%[8]、58.10%[10]、61.25%[11]、64.0%[9]),而与既往过敏史的相关性,报道不一。儿童ADR一般较轻微,99%以上好转或治愈。国内外报道均类似。
3.药品说明书
药品说明书长期存在格式不统一(纸张、字号太小等)、名称项目标准不统一,表达不明确,不同厂家撰写的说明书内容不统一,差异较大等问题。缺项现象普遍存在,其中[药物过量]、[儿童用药]、[老年用药]、[孕妇及哺乳期妇女用药]、[药物毒理]、[药代动力学]各项缺失率较高,中药缺项率远远高于化学药品和生物制品。进口和合资药品说明书内容比较直观、详细清楚,缺项情况好于国产药品,但进口药品试验用人种不明确[13-16]。
[儿童用药]缺项率高,但各文献报道不一,缺陷率范围为14%~47.4%,进口和合资药品此项缺失率较低(5.7%,8.3%),国产药品此项缺失率相对较高(37.3%,45.0%)。中药儿童用药的标注率极低,缺失率极高(55.9%、64.7%、100%),外用药缺失率也高。综合性医院用药说明书此项缺失情况较专科医院严重。说明书中表达过于笼统,多见“暂无儿童用药资料”、“儿童用药的安全性和有效性尚未确定”、“儿童用量酌减”、“慎用或遵医嘱”等模糊词句,未标明用法和用量。口服药品是否可与牛奶、果汁同用,用药前有何注意事项,药品的服法、治疗周期、是否禁食、是否需分剂量、是否可中西药合用等一些细节问题很少提及。另外,还存在儿童药物剂量计算方法无统一规范、标准,不良反应、药代动力学项下多未提及等问题。无儿童剂量或儿童用药说明比例较高的是眼科和皮肤科用药,较低的是呼吸系统药物[17-26]。
(三)我国儿童用药的现状及问题
我国儿童用药存在儿童专用药物品种、规格、剂型少的现状。儿童用药药品说明书中,[儿童用药]专项的缺失率仍较高,尤其突出在中成药中,且儿童相关信息表达含糊、不规范现状较为普遍。如何加强、完善儿童用药药品说明书的监管应是目前的切入点。儿童药物不良反应多见于抗感染药物,这与儿童用药的特点相关,规范抗感染药物的说明书、提高其合理使用的水平应优先考虑。
三、儿童用药情况上海地区的实证研究
(一)实证研究的目的和方法
为提高课题研究的客观性和科学性,采用观察法、谈话法、测验法等实证研究的方法,观察调研上海地区儿童用药的实际情况,以期通过对上海地区儿童用药现象的大量观察、调查和分析,获取客观材料,从个别到一般,归纳出我国儿童用药的基本现状及存在的问题。
(二)调查问卷的设计、收集与整理
调查问卷涉及目前儿童就医情况,医院规章制度,药品规格、剂型、说明书,不良反应监测系统,儿童用药法规政策等方面。对上海市400多家医院院办、儿科和药房进行了问卷调查。共发放1 400份“医院儿童用药情况问卷”,回收189份,其中完整的为170份、退回5份。有效问卷中儿科专科医院84份、综合医院有儿科50份、综合医院无儿科24份、其他26份,其中儿科专科医师62份。对于目前儿科用药存在的问题,专业人士认为排第一位的是用药制度不健全(66.8%),其次为说明书不规范(60.3%),认为药品品种少、剂型少和规格少的分别占48.4%、40.8%和41.3%。参与问卷调查的专业人员基本上都认为建立儿科规范给药的用药程序或制度是有意义的,但仅56.7%的医院设立了相关制度。99.4%的认为开发儿童专用药品是必要的,97%的认为建立儿科基本药物目录、公开儿科用药信息和出台规范儿科用药的相关法规、政策必要且重要。目前绝大部分医务专业人员(88.3%)对不良反应上报系统已有一定的认知度和熟悉度,仅8.2%的人员对此系统不熟悉,还有3.5%的人员不知道。值得注意的是,对于在儿童群体进行临床试验是否可行存在一定的分歧,返回问卷中69.6%认为必要,30.4%认为不必要、不可行,而儿科专科医师中96.8%认为儿童群体临床试验是必要的,见图1、图2。
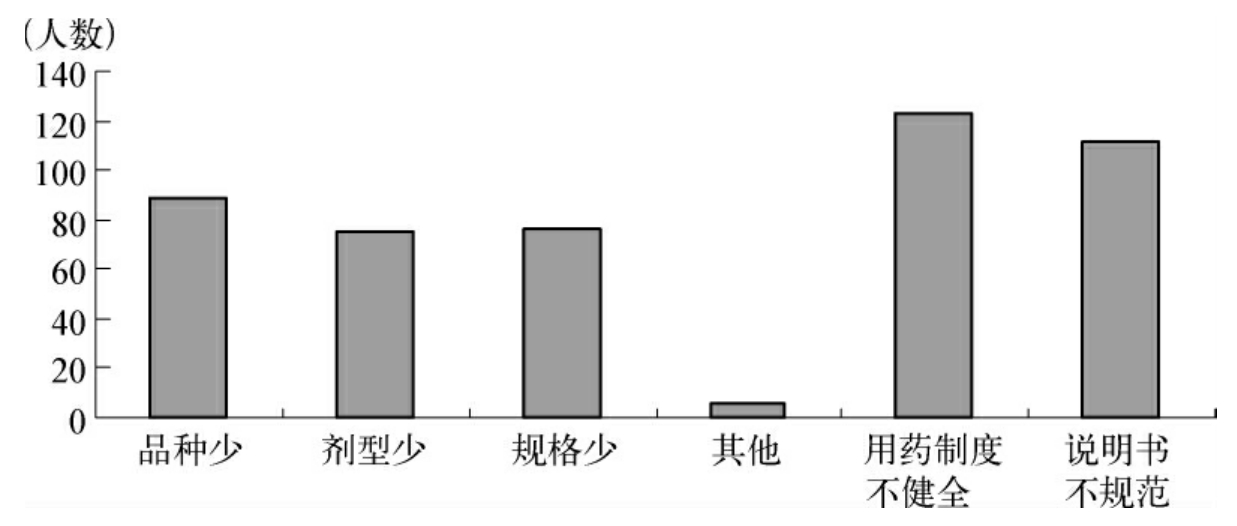
图1 上海地区专业人士认为目前儿童用药存在的问题

图2 上海地区专业人士对儿童用药有待解决问题的看法
(三)从上海市不良反应监测结果看儿童用药
分析“上海市药品不良反应监测网数据库”信息,从2003年底~2009年底该数据库共收集儿童ADR报告8 299份。患儿中男性占58.59%、女性占41.41%;多为一般报告(97.48%),严重报告占2.52%;3岁以内儿童ADR发生率高(见表1)。易引起ADR的药物中抗感染药居前,疫苗引起的ADR亦较多(见表2)。以静脉给药方式引起ADR最为多见,肌内注射次之(见表3)。在所报告的总计10 300例次的不良反应中,ADR多见皮疹、发热(见表4)。大多转归良好,治愈+好转占99.39%。
表1 上海市儿童药物不良反应年龄分布
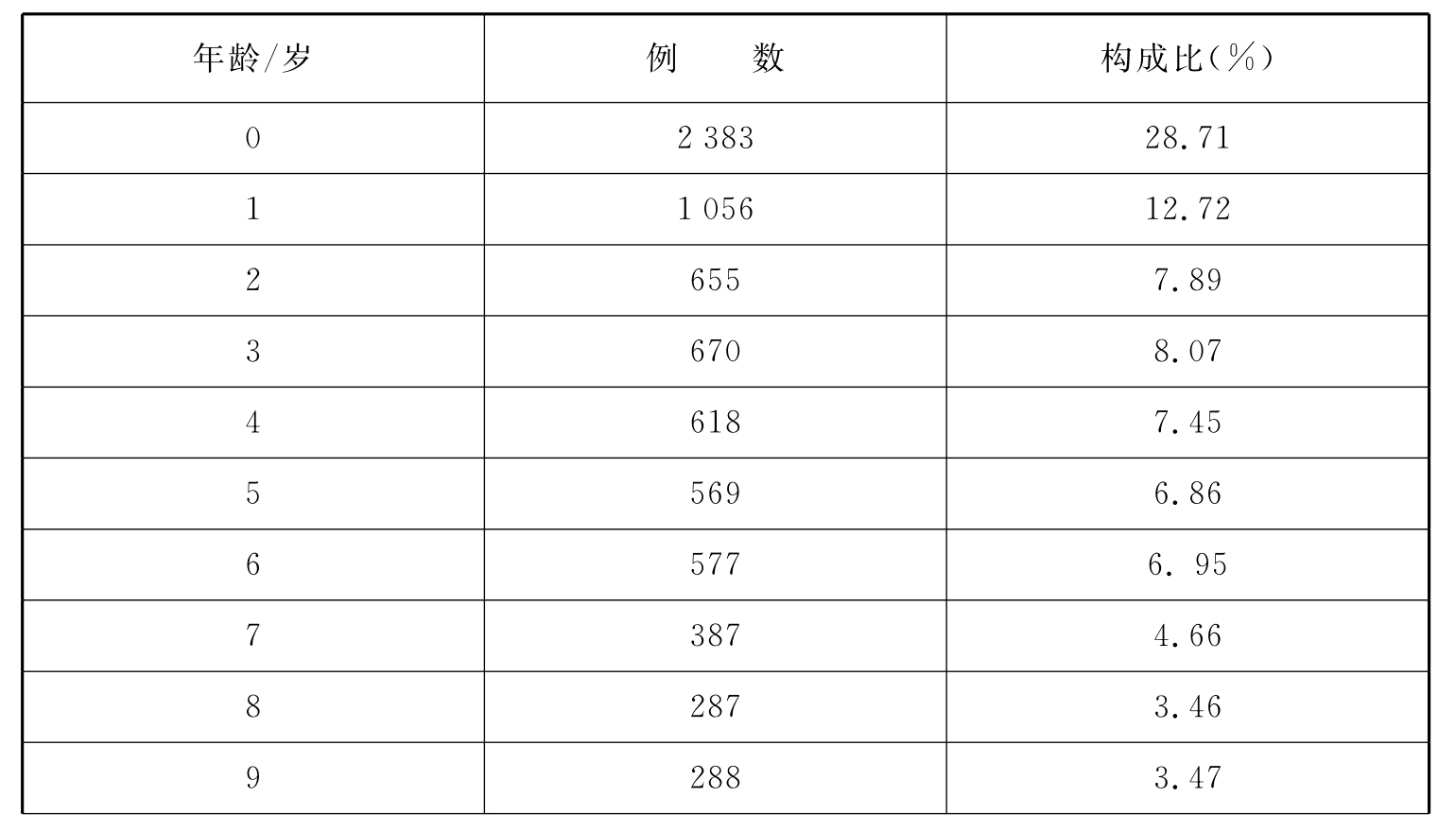
续 表
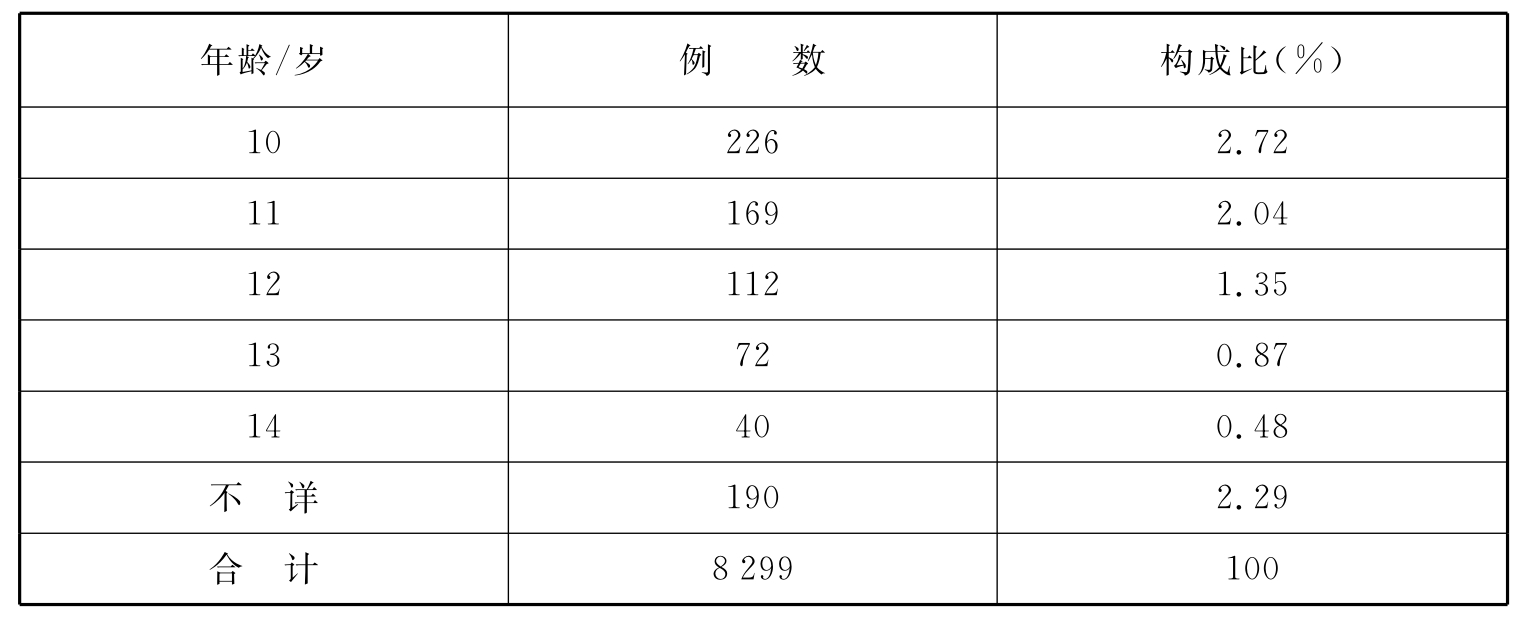
表2 上海市儿童药物不良反应常见药物
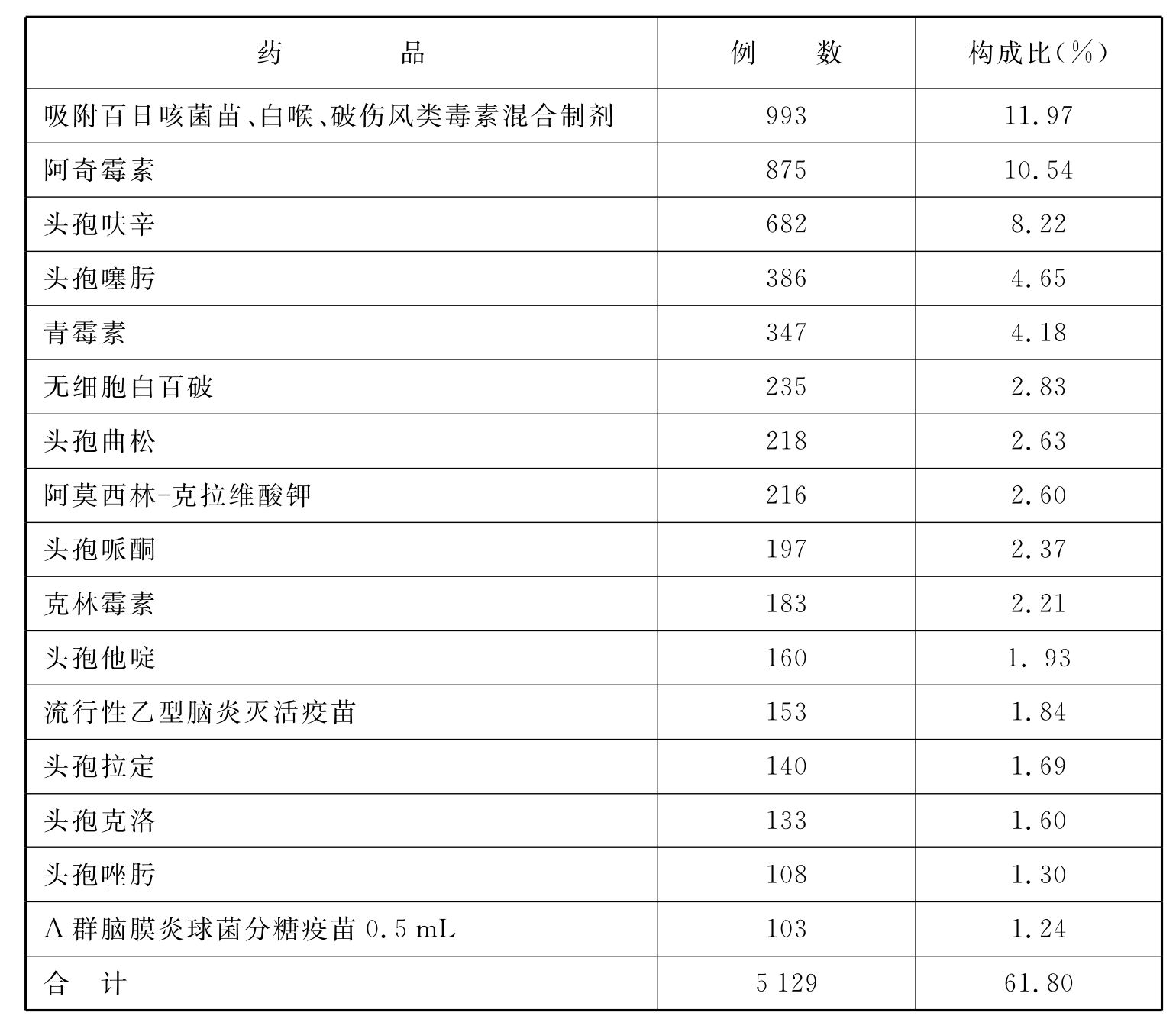
注:所列为前16位,100例以上的不良反应
表3 上海市儿童药物不良反应用药方式
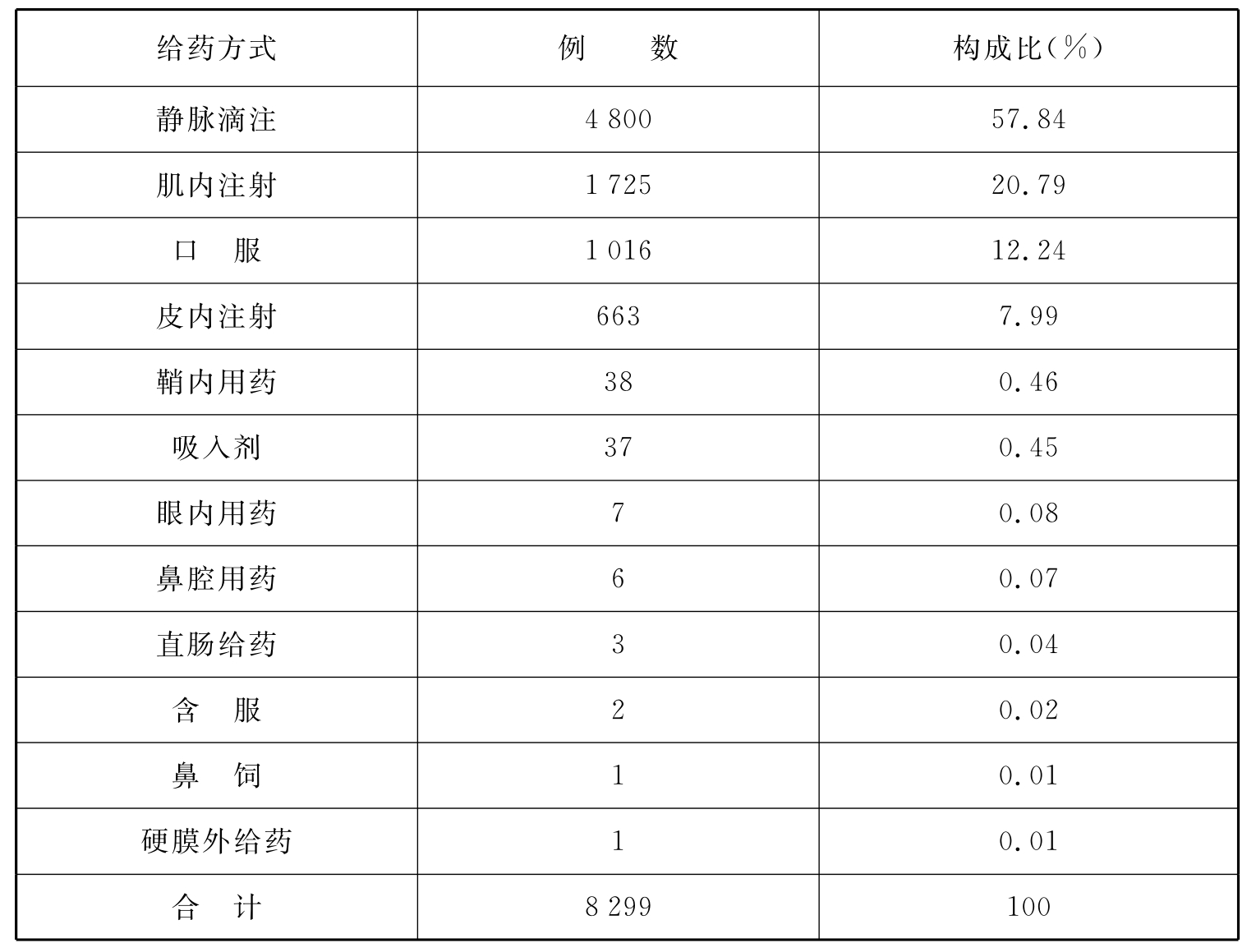
表4 上海市儿童药物不良反应类型
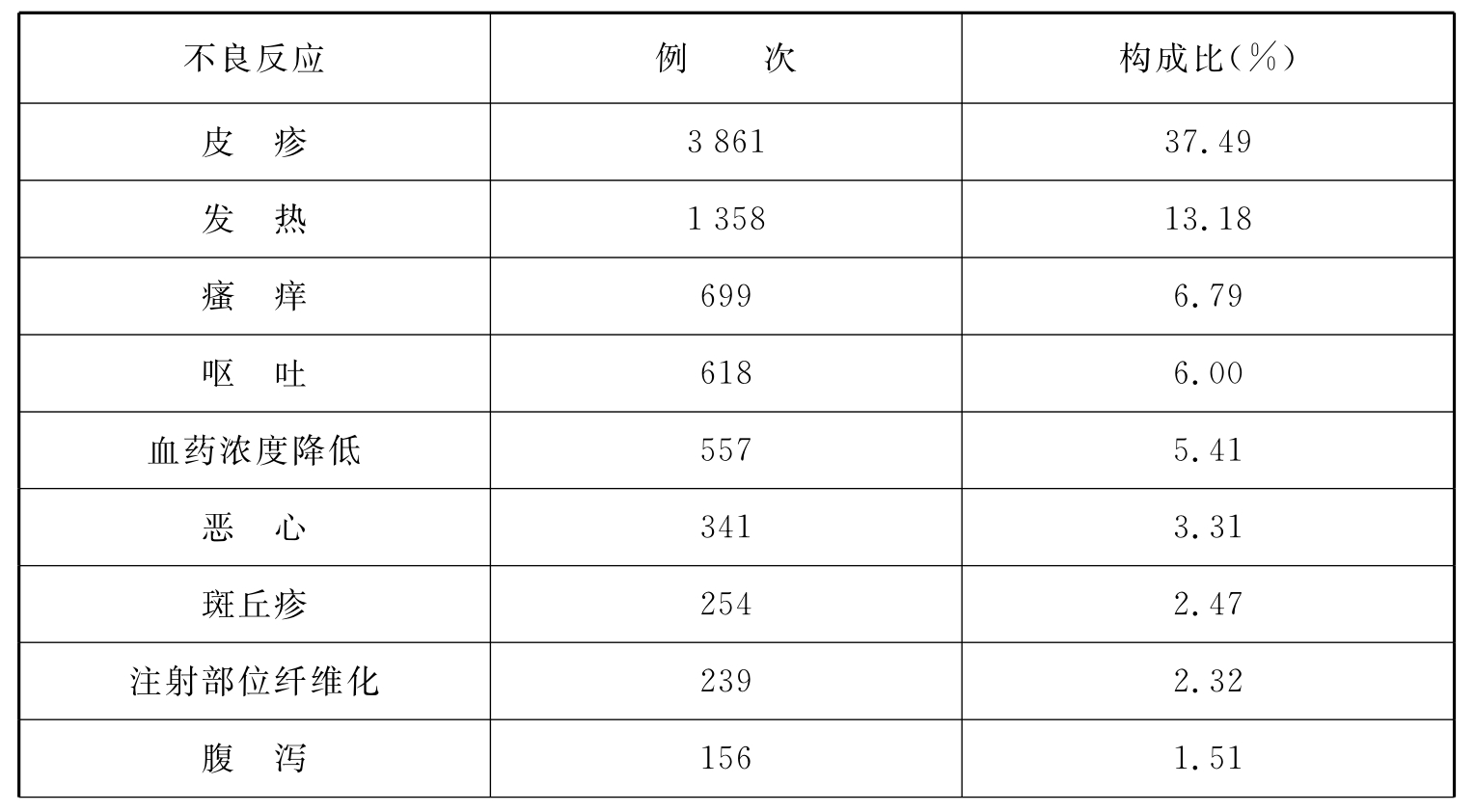
续 表
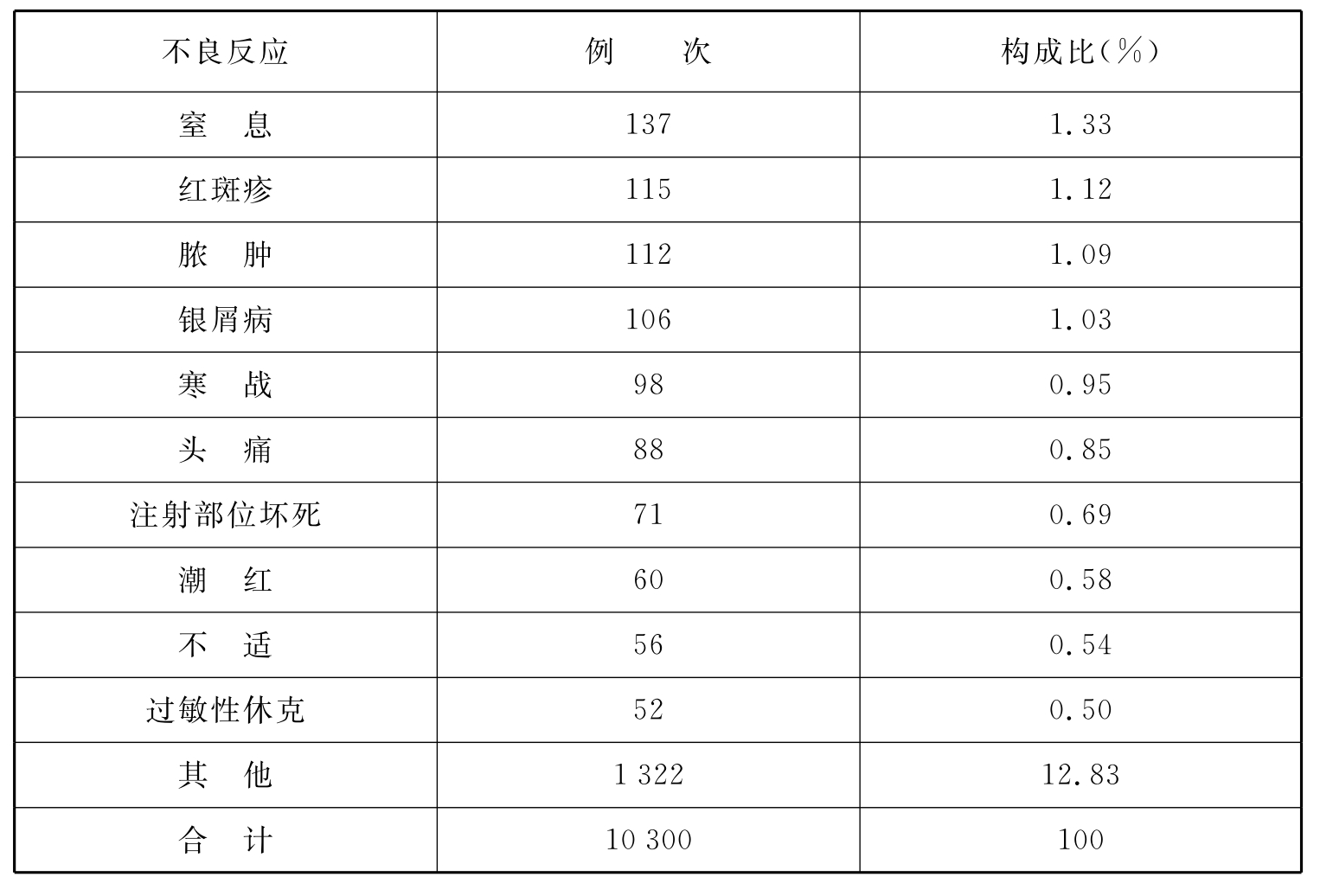
(四)从长江流域医院用药信息数据库看上海儿童用药
按药品通用名检索“长江流域医院用药信息数据库”,分析儿童用药(包括适宜儿童用的剂型如咀嚼片、喷雾剂、混悬剂、颗粒剂、滴剂、口服液、糖浆、泡腾片以及药名中标识小儿、儿童等特征的药品,不包括针剂)在上海地区两家儿童专科医院中的使用情况。
通过对此数据库分析,上海市两家儿童专科医院2008年用药总金额1.89亿元、575种药品、908种品规,其中:儿科药物总金额0.71亿元(37.6%)、127种药品(22.1%)、171种品规(18.7%)。2009年1~10月用药总金额1.81亿元、573种药品、886种品规,其中:儿科药物总金额0.64亿元(35.3%)、132种药品(23.0%)、169种品规(19.1%)。
(五)从专家访谈看上海儿童用药
采取走访和电话联系的方式,对儿科专科医师、专科医院药师、药企儿童产品专员等进行访谈,了解上海市儿童用药情况、存在的问题以及专业人员对现状的看法。
儿童用药目前的确存在诸多问题。儿科病人中代谢性疾病逐渐增加,药物治疗非常重要,但专门的儿科药物非常少且较多的成人药品不宜在儿科使用。儿科医师如何处理此类问题,应该有制度,对特殊病例特殊处理。针对少见病和疑难病,需要有特殊用药指导、途径和药源。
儿童实际用药品种太多、乱用,同一药品商品名过于复杂,儿科药物的剂型、规格少,口感太差,减少分药现象非常重要。价格便宜的药逐渐淘汰,价格较贵的药品上市,但两者效果相似。应开发儿科病人专用药物剂型,符合儿童口味,以液剂较好,便于服用,且可小剂量使用。造成目前儿科用药品种少、剂型、规格少的原因很多,但经济因素是最重要的一环。由于儿科用药量少、要求高,利润也就相对低,因此研发、生产、流通单位积极性不高。政府必须从经济政策上扶持。最好要有正规儿童用药专业厂家生产药品,适当改善药品口味,做到剂型多样化,经济实用。
药品说明书中儿童用药方面资料不全,儿科用药用量、用法不健全;儿科医院自制制剂缺少具体用法用量、不良反应及注意等事项说明,用药剂量不规范、缺少用药安全性数据。药物说明书中不良反应发生率多少应写上,避免患者看了说明书害怕药物不良反应发生。药品说明书的制定应措辞应严谨,避免不必要的矛盾。对在儿科中应用的中成药说明书须加以修改,在严格规范的临床试验基础上,可参照某些西药,根据年龄及体重计算药品服用剂量。首先要有严格的制度,切切实实把好“国药准字”的审批关,对审批机构和人员要有问责制,对批准的每一项新药的各项质量指标均应认真把关。医院进儿科药品时,生产厂家要有一定知名度、一定规模,以确保临床用药的质量和安全。每种药物均需专业单位质量控制证明,静脉用药尤其是中药注射剂使用时更要谨慎。
儿科患者市级专科医院就医人数明显增长,社区一级医院减少,而儿科用药问题多出现在基层医院、外地患者。社区卫生服务中心是卫生保健的基础,但目前是儿科最薄弱的环节,很多地方甚至还是空白。越是大医院,儿童用药的档次越高,医生为了能让家长迅速看到治疗效果,减少医疗纠纷,尽量采用效果好、价格高、级别比较高的品种,价格贵不说,对儿童的体质、用药的效果及抗药性、药品耐受性都不利,希望能对医生及儿童家长做好相关宣传和教育工作。另外,儿科呼吸道感染等许多自限性疾病无需抗生素治疗,而家长及部分医生给予抗生素治疗,反而影响小儿生长发育及健康。总之,应让广大群众了解规范儿科用药的重要性,应加强儿科用药的公众宣传,对家长进行用药培训,提供咨询。
在医院门诊与病房增设相关临床药师,及时或实时收集儿科用药安全与不良反应的资料。建议对儿科临床药师多加培训,发药同时讲清药物的主要不良反应。建立临床药师下病房,与临床医师共同研究疑难病用药。建立常用儿科药物的药物浓度监测,指导临床药物的使用。将药师的儿科用药调配作为业务考核的重要内容,纳入药事管理。有专家指出,儿科用药均应建立在儿童群体临床验证基础上。但目前国内儿童临床试验基地少、志愿者少,而且医生临床试验经验少、不专业。成人用药在进入儿科临床前也应进行相应临床试验,包括中药。
儿科用药量应按体重计算,区别同龄个体差异。医生开具的处方大多数情况下“一方多用”,应根据患儿具体情况增减用药。加强对临床医师所开处方的审核和考量,加强儿童用药的再培训和信息更新。儿科用药知识来源尚未系统建立,应定期对医务人员进行儿科用药的知识培训。应规范用药,减少静脉用药,严格控制儿科中药制剂的应用。
建立高层面的儿科用药督查制度。规范儿科用药的相关法规、政策很重要。相关部门应联合中华儿科学会各学组制定相关用药安全、有效制度。医保目录的品种少,应完善儿科用药基本目录。建立管理科学的诊疗用药体系和费用指标体系,建立统一的、科学的儿科绩效考核章程,保障综合医院儿科医师待遇。建立切实有效的政府对综合医院儿科的投入机制,建立规范的儿科药物推广招标制度。
(六)上海地区儿童用药基本情况
上海地区专业人士对儿童用药的关注度是极高的,普遍认为目前存在一系列问题有待解决和改善。儿童用药制度的不健全和说明书的不规范是最受关注的问题,出台规范儿童用药的政策、法规得到绝大多数专业人士的赞同。目前绝大部分医务专业人员对不良反应上报系统已有一定的认知度和熟悉度,值得注意的是,对于在儿童群体进行临床试验是否可行存在一定的分歧。儿童专用药品的品种、剂型和规格仍偏少,在上海市儿童专科医院中儿童专用药品的份额仅25%左右。药物不良反应最常见于抗感染药物,这也是合理用药的薄弱环节。加强社区儿科建设、提高临床药师儿童用药水平并多参与临床工作是今后注重的方向。
四、国内外儿童用药管理比较研究
在我国,目前相关法规比较缺少,而以美国为代表的发达国家通过立法逐步建立了较为完善的儿童用药管理制度。在这些法规中“儿童独占”制度的提出,受到大家的注重并已取得较好的社会、经济效益。
(一)美国儿童用药管理
1.概述
在美国与儿童用药相关的法律中最主要的有《联邦食品药品化妆品法案》(FD&C)及《儿童最佳药物法案》(BPCA)和《儿科研究平等法》(PREA)。BPCA是在2002年1月4日颁布的,是FD&C的重要补充法案,包含5年的日落条款,2007年经修订后继续生效[27,28]。
2.组织管理机构
FDA设有儿科疗法办公室(Office of Pediatric Therapeutics),此办公室统管儿科的伦理学、儿科药品上市后研究以及对儿童用药安全知识教育等一系列涉及儿科用药安全有效的问题。另外,FDA设立了两个与儿科用药安全相关的委员会——儿科咨询委员会(PAC)和儿科伦理小组委员会,对FDA的儿科药品问题提出建议。
公共卫生和科学办公室以及食品药品委员负责《联邦食品药品化妆品法》、《儿童最佳药物法案》、《儿科研究平等法》等多部公共卫生服务法的实施。儿科咨询委员会就儿科治疗、儿科研究以及其他任何FDA监管职责内与儿科有关的事宜,为FDA食品药品委员提供咨询和建议。此外,委员会也就儿童参与研究的项目,为公共卫生和科学办公室提供咨询和建议。该委员会一年开3次会议。
儿科伦理小组委员会是伦理委员会的一个分会,探讨儿科研究的伦理问题。例如,成人能够对参加临床试验给予知情同意(informed consent),而儿童不能,因为该“同意”(consent)意味着完全理解潜在的风险和其他因素。父母被陷入让孩子加入某一研究的决定之中,并且7岁或更大一点的孩子可能“同意”(as-sent)或“不同意”(dissent),意味着他们可能同意或不同意参加某一研究。
3.儿童药品独占权[28-31]
1997年在美国通过的《食品和药品现代化管理法》(FDAMA)中首次制定了儿科独占条款(Pediatric Exclusivity Provision),为鼓励制药商进行儿科临床研究,允许授予6个月的儿科市场独占期。2002年BPCA对儿科独占条款的相关内容进行了修改和完善,再次鼓励制药商对儿童群体进行临床研究。
儿科独占的获得必须经过一系列完整的程序。首先是FDA向药品申办者发放书面请求,药品申办者根据书面请求对儿科研究的要求对FDA做出回应,如果该药品申办者同意进行儿科研究,就和FDA就儿科研究的具体细节签订书面协议。药品申办者按照书面请求和书面协议中规定的期限完成儿科研究要求的所有研究项目,并以新药申请(NDA)或补充申请的形式向FDA提交儿科研究的研究报告,如果该研究报告的内容与书面请求和书面协议的要求完全相符,FDA就会授予该药品6个月的儿科独占保护期。
FDA和美国国立卫生研究院(NIH)会制定一个年度药品目录,将一些没有(或已失去)专利或其他独占保护的已被批准上市的药品列入名单。之所以将这些药品纳入药品目录,是因为FDA和NIH认为,这些药品在儿童中使用需要进行额外的儿科研究,以获得这些药品在儿科群体中应用的有效性和安全性信息,并且对于列入药品目录中的药品,FDA将会给该药品的所有持有者(包括原专利或独占保护持有者以及仿制药持有者)发放书面请求。对于还未被批准上市的新药,以及还有专利保护或独占保护的药品的持有者,需要向FDA主动递交提议的儿科研究请求(PPSR),以获取FDA发放的书面请求。如果专利药品或独占保护药品的持有者不主动获取书面请求,而FDA和NIH认为该药品需要进行儿科研究,则该药品也可能会被纳入药品目录。另外,其他对儿科临床研究感兴趣的第三方非药品持有者,也可以向FDA递交PPSR,以获取该药品儿科研究的书面请求。对于药品主办者,是否接受书面请求是自愿的。儿科独占期为6个月,但6个月的独占期生效的前提条件是该药品是经FDA批准上市的,并且必须续加在该药品列入桔皮书的现有的专利保护期(包括延长的专利保护期)或其他独占保护期(如数据保护、罕用药独占和仿制药独占)后面生效,是相当于对原有专利保护或独占保护的延长。
一份书面请求只能获得一次儿科独占。但是,如果在获得儿科独占后,药品主办者又收到了一份不同的书面请求,并且按照新的书面请求的要求完成了儿科研究,则药品主办者可以获得第二次的儿科独占,只要满足以下条件:按照新的书面请求进行儿科研究的结果获得了该药品的儿科新用途(包括增加新的儿科适用群体);这个新用途未在该已批准的药品的标签中标注;增加的新用途还使得药品满足Hatch-Waxman法案的要求,获得3年的数据保护;6个月的儿科独占附加在,并且只附加在这3年的数据保护之后,而不能延长其他的专利保护或独占保护。儿科研究的实质是对药品中含有的某一种或几种有效成分在儿科人群中应用的安全性和有效性研究。因此6个月的儿科独占仅续加在针对这一种或几种有效成分的专利保护和其他独占保护期后生效。如果药品主办者同时拥有多个药品都含有这一种或多种有效成分,则每一个药品都可以获得6个月的儿科独占。
4.儿童药品研究管理[32-34]
在20世纪90年代早期,FDA执行自愿的儿科研究政策,但它们大都是不成功的。1997年,FDA公布了一个被提议的规章,首次要求新的药物和生物制品的制造商在某些情况下进行儿科研究。该规则于1998年被定案,第一个研究被要求于2000年12月开始提交。但是,该规则遭受了批评。2000年12月,美国内科医师和外科医师协会、竞争企业研究所和《消费者警报》起诉了该儿科规则,挑战FDA要求儿科研究的法定权力。2002年的10月,一个联邦地区法院推翻了该儿科规则。
2002年12月中旬,健康与人类服务部(HHS)部长宣布,HHS将推进法案的通过,该立法将赋予FDA要求制药商进行适当的儿科药物临床试验的权力。2003年,美国国会通过了该法案,在法案中制定了强制性儿科研究的儿科规则。儿科独占和儿科规则共同作用,被形象地形容为“胡萝卜加大棒”儿科研究。不同于独占权条款,儿科研究规则是一种要求,并适用于药物和生物制品——来源于诸如疫苗、血液和血液衍生物等活体来源的医疗产品,以及癌症的新治疗剂。通过这一规定,在药物的早期开发中就应将儿童用药考虑在内。
儿科研究规则被计划应对儿科独占权条款留下的某些漏洞。根据儿科规则,如果FDA确定该产品可能被用于一定数量的儿童患者,或者如果该产品对儿科人群将提供的有意义的利益超过现有的治疗时,FDA可以要求某种已提交新药申请的药物进行儿科研究。当然,儿科规则并不延误成人用药物的获得。
《儿科研究平等法》规定针对新的有效成分、新的适应症、新的剂型或新的给药途径等提出的新药申请或补充申请必须进行儿科评价,除非有合适的理由经过FDA的同意准予延迟或放弃。儿科评价是指对药品在相关儿科人群中针对申请批准的新的适应症应用的安全性和有效性评价。这与儿科研究的要求有所不同。儿科研究要求进行的是对药品中的有效成分在所有儿科群体中针对所有适应症应用的安全性和有效性研究,无论这个适应证是否在成人中已经通过了批准或还未通过批准。因此,虽然儿科规则对一些药品做出了强制性的儿科研究,但它对儿科研究的要求与儿科独占是有区别的。仅仅按照《儿科研究平等法》的要求完成儿科评价只能够被批准上市,但不一定能够获得儿科独占。必须按照上文所述,向FDA递交PPSR,以获取书面请求,并全面完成要求的儿科研究,才有资格获得儿科独占。并且,儿科评价必须在药品批准上市前进行并完成,而儿科研究既可以在药品批准上市前,也可以在上市后进行或完成。所以,儿科独占和儿科规则是两项相互补充,但又各自独立的政策。
5.儿童用药信息公开
无论是否可以获得6个月的儿科独占,在研究结果备案后的180天内,FDA都会将所有研究信息(包括药品主办者的信息、药理学评价信息和所有的临床研究信息等)在《联邦注册》杂志上公开发表,该杂志可以在FDA的药品评价中心(CDER)网站上在线获得。儿科评价的研究信息在FDA的网站上公开发布,并且在药物标识中标明儿科信息,否则也会被认定为错标药物,并从市场中召回。NIH和儿童健康和人类发展部(NICHD)还联合成立了儿科药理研究单位协作网,有多家儿童专科医院协作参与,公布、探讨儿童临床研究的最新进展。
儿科研究备案后,该药品所有者应该修改药物标签,FDA给药品持有者180天限期与FDA达成标签修改的协议。如不能达成协议,则交由儿科咨询委员会与药品持有者协商,以最终达成协议。如药品持有者仍坚持拒绝修改标签,FDA会认定该药品为错误药物。另外,FDA规定在获得儿科独占的1年后,药品主办者必须向儿科疗法办公室报告该药品所有的不良事件信息。
药品说明书是药品信息的主要载体之一,药品说明书的监管非常重要。美国FDA在药品标签和说明书的监管方面积累了大量成熟的经验,与药品标签和说明书相关的法律文件大致分为3类:法律(act)、联邦法规(regulation)和指导原则(guidances)。其中,法律和联邦法规是企业必须遵守的法律要求,具有法律强制性;而针对企业的指导原则属于建议和指导性质,供企业参考使用。现行相关法律包括《清洁食品和药品法》(1906年)、《联邦食品药品化妆品法》(1938年)、《Durham-Humphrey修正案》(1951年)、《Kefauver-Harris药品修正案》(1962年)、《正确包装和标志法》(1966年)、《处方药市场营销法》(1988年)、《食品和药品管理现代化法》(1997年)、《人用处方药品及生物制品说明书格式及内容管理条例》、《人用处方药品及生物制品说明书新内容与格式管理条例实施指导原则》、《人用处方药品及生物制品说明书[临床研究]内容格式指导原则》、《人用处方药品及生物制品说明书[不良反应]内容格式指导原则》、《人用处方药品及生物制品说明书[注意事项]、[禁忌症]、[黑框警告]内容格式指导原则》。联邦法规主要是指美国《联邦法典》(CFR),药品部分主要集中于HHS颁发的第21篇(简写为21CFR),药品标签和说明书的相关法规收编于21CFR1210和330。指导准则分为正式(final)和暂行(draft)两种,内容主要是关于药品标签和说明书的内容与格式、特殊药品的标签管理等。2006年1月18日发布了的《人用处方药品及生物制品说明书格式及内容管理条例》,旨在完善处方药品说明书格式,突出说明书中最重要的信息,提高清晰性、易读性,便于查询。新增了概要、目录两部分新内容。概要位于药品说明书首页最前部,一般为半页纸,可让医生对药品的利弊信息立即得知,并一目了然。此栏目信息对于医生开具处方是最重要的,它精确概述了黑框警告、作用与用途、剂型与给药途径、用法用量等项中的主要内容;并且要求注明各项内容在说明书正文中所处的条目和位置,引导医生去阅读更详细信息。本栏目中,药品生产企业必须列出本年度内该产品的实质变更项目,使医生在开具处方前了解产品最新信息。目录可方便医生快速查找并锁定某项内容,医生可根据目录标注条目号,在全说明书中查阅详细信息内容,同时使说明书的条理性更加清晰。另外,还增加了初始批准日期、免费联系电话及网址、患者忠告等项目[35,36]。
目前儿童用药领域,未获得许可(unlicenced)及超说明书(off-label)使用非常普遍,在欧美等发达国家也存在这样的现象,50%以上的药品在尚未获得充分的儿童研究资料的情况下在儿童中使用。unlicensed用药包括许可的药物再调配,药物许可、但剂型未许可的,特殊生产许可范围的药品,药物在另外国家许可、但在本国未取得许可证等。off-label用药是指药品使用的年龄、适应证、给药方法、剂量、给药途径不在厂家提供的药品说明书之内的用法。这种用药方式可造成疗效不确定、用药剂量偏高或偏低、影响儿童生长发育等,同时会显著增加与药品相关的不良反应发生率[37,38]。
(二)欧盟儿童用药管理[39-43]
1.概述
2007年1月26日《欧洲儿科药物规则》生效,这在欧盟是一个变革,旨在激励适用儿童年龄药物的研发,提高儿童用药信息的可信性和可利用度。首先,企业被要求在儿童人群中作药物研究,开发与各年龄相适应的药物配方。作为激励或回报,企业有资格延长专利保护或获得市场独占权。这一规则要求建立一个欧洲儿童临床研究网,为专利到期的药物构建了一个儿科研究方案,后者可通过欧共体框架计划获得资助。儿科药委员会,隶属于欧洲药品评价署,负责评审企业的儿科研究计划(PIP)。儿科研究计划描述在儿童群体中研发一种药品进行临床试验和其他一些相关措施的必要性。欧洲儿科临床试验数据库中包含与儿科研究计划相关的临床试验的详细信息和结果,会上网公布,其中一部分公众易获得。所有获得儿科使用许可的,会在包装标签上有一个新的标志予以区别。5年后,对此规则重新评价,看是否需要修正。
2.组织管理机构
《儿科药物规则》规定在生效6个月内成立一个新的科学委员会——儿科药委员会(PDCO),于2007年7月26日成立。PDCO成员包括人用药委员会(Committee for Human Medicinal Products,CHMP)中的5个成员及他们的候补人员。CHMP是EMEA中主要科学委员会,负责对医药产品上市许可的申请给予评价,并对药品的发展提出科学的建议。这5个成员是2个委员会之间的重要纽带。
不能由任一CHMP委员体现的成员国可以在PDCO中指定一名委员及候补委员。另外,欧共体会指定3个委员及其候补委员作为健康专业人员,3个委员及其候补委员作为患者联系人。欧共体会指定这6个委员及其候补委员关注公众关注的事情,并反馈至欧洲议会以供参考。这些任命都是希望通过合作,确保委员会的最终组成(包括委员及其候补人员)覆盖儿科用药的各个相关科学领域。委员只有在委员会会议中被认定为其他相关领域的专家,才能再任期3年。这些委员、候补人员及专家必须与制药企业无任何经济和利益上的联系,以保证他们的公正性。
PDCO有许多重要职责,他的有效性是《儿科药物规则》成功的关键。其最主要的职责是评估PIP内容并给予判断,以及对申请作出豁免或推迟的决定。在这一点上,如果PDCO考虑到一种药品在儿科领域应用不安全、疗效差,或是不能提供显著的治疗益处,那么PDCO可强制豁免。PDCO也可以应要求,对已获得PIP批准的上市许可申请的可行性及一种儿科药物的安全、质量和有效性作出评估。PDCO还有一些特殊的任务,如建立儿科特殊需要用药的详细目录、收集输入与实现规则目标相关的前沿文献等。
PDCO同时是EMEA和欧共体的顾问,在一些问题中提出建议和意见,如在所有确认可在儿科使用的药品标签上作新的标志、建立欧洲儿科临床试验网、如何交流在儿童人群实施相关研究,以及其他与儿科用药相关的问题。
委员会每月召开3~4天的会议,委员们在会前要用大量的时间作准备,所以全体委员的工作量是不能低估的。
3.儿童药品独占权
以前药企只被要求做成人群体药物的研发,儿科药物的研发带有随意性。这个规则是欧洲法律中第一次要求药企研发在一特殊患者群体中使用的药物。因此,此规则中最重要的要求就是要求所有申请上市许可的新药,包括罕见药,必须具有先前批准的PIP中所有研究的结果和信息。
PIP应包含所有研究及时间节点的详尽提议,以支持这一产品在儿科中的使用,也应覆盖所有的儿童年龄段及必要的与年龄相适应的配方。这是规则的缺陷所在。在一些情况下,这会使成人中相关产品的研发部分或全部延迟,这时有关的儿科数据可在最初的市场许可后提供。在另外一些情况下,这一要求在部分或全部儿童中豁免,例如,在没有儿科治疗需要或儿科使用这种产品是不合适的情况。但是,最重要的一点是,不管是延迟还是豁免,对于一个新药来说,在申请许可时必须提供儿科数据。
这一要求还应用于受专利保护的药物添加新适应症、新药物剂型和新的使用途径。此规则还包括激励企业开发儿童使用的药物。这不但指激励以上提到的研究规定的儿科产品开发,还包括给药企实行必要的研发程序所需的奖赏。
当一个批准的PIP完成,所有的信息呈递给管理部门后,这个药品可以得到额外的6个月专利保护期(是对其药品补充保护的扩展),并且这个药物在所有成员国都会被许可,其相关儿科研究信息,不论是正面的还是负面的,都在产品特性摘要中整合出来。无论资料是否支持儿科适应症,这一延长都会被允许。激励只会在有有效药品补充(SPC)保护的产品中产生,无SPC或SPC过期的产品则得不到经济奖赏,同时没有机制可以延长专利保护期本身。对于罕见病药,激励机制可使其10年的市场独占期再延长2年。
经济激励不会回顾性给予,因此,规则强制执行以前完成的PIP内容得不到奖赏,除非PIP中有极其重大意义的研究,且是在规则强制执行后完成的。
要求及激励机制是相互独立的。因而,一个获得市场许可者会被要求提供一个完整的PIP,而没有资格获取奖赏,比如,因为在PIP完成前SPC过期了。相反,一个不需要呈递PIP完整申请的市场许可获得者,为了从激励机制中获得益处而提出PIP申请。
规则为了激励专利保护到期的药物在儿科中使用,还建立了一种新的市场许可,称为儿科使用上市许可(PUMA)。只有儿童专用药品才有资格申请PU-MA。与PUMA相关的激励是相对薄弱的。但是,仍希望通过PUMA能鼓励从那些老产品上开发出新的儿科配方药物。PUMA申请必须包含已批准的PIP结果,可参考相同活性成分的第三方资料,能使用集中许可程序,一旦获得许可,可获得10年市场保护期。
4.儿童药品研究管理
儿科研究计划(PIP)是为保证一个药物能被批准在儿科应用提供必须数据所制定的研究和开发方案。对于新药上市批准、新的适应证、受专利保护的已批准药物新剂型或新给药途径,以及提交PUMA申请,都必须事先拟定PIP。PIP必须详细阐明研究方法和时间安排,以保证药物在所有相关儿科群体中应用的质量、安全性和有效性(ICH E11中规定的)。另外,PIP中必须描述如何使不同的儿科群体获得更易接受、安全、有效的用药配方。
为了确保在全面的产品研发中儿科用药部分是事先很早阶段就考虑到的,而不是事后产生的想法,PIP必须在成人药动学试验完成之前提交到儿科药委员会(PDCO)。这导致在许多案例中,最初的递呈在研发的很早阶段产生,非常初级,以致不能提供一个完善和详细的计划。因此,《儿科药物规则》允许PDCO要求研发方对计划提供进一步的信息或进行最初的修改,也允许在产品研发过程中对计划进行修改。但是,为了达到上市许可有效申请所要求必备的条件,并达到具有符合规则提供的财政激励标准的资格,计划必须经PDCO认可。
如果PDCO拒绝认可一项PIP,申请者可以要求PDCO进行重审。如果接下来的重审,委员会仍然给予否定,而申请者不同意这一评价,可以向欧洲法院申诉。同时,对申请人提出的豁免或推迟PIP请求,PDCO将予以批示。
儿科研究方案对专利到期药物或是活性成分在儿科中使用的研究的资助要由社区研究计划提供,确切的资助数目在规则中没有详细标明。这个是在欧洲议会、各成员国以及科学团体、企业和其他资助者共同协商的基础上,由欧洲议会建立的。
为了在规则支配的框架计划下获得社会资助,申请者必须充分阐述研究的必要性,以成功投标。研究必须在欧盟中一个以上的成员国中进行,或者具有资格的相关候补者或第三国家。PDCO会编制一部详细的儿童领域治理需求的目录,以帮助明确研究重点。
5.儿童用药信息公开
这一规则同时要求EMEA为儿科临床试验开发一个网站,这个网站可以把现有的与儿童群体研究相关的国家和欧洲网站联系在一起,把个体研究者和具有特殊专业的中心联系在一起。这一欧洲网的目标是调整儿科药物相关研究,建立必要的欧洲范围的科学标准、管理标准,避免在儿童群体开展不必要的、重复的试验和研究。
虽然对于这个网,规则没有指定特殊的资助,但是EMEA的预算中有一些是可以用到的。子网站、中心研究人员为了自身的基础设施和研究活动还需要寻求其他来源的资助,比如中央政府、药品企业或者欧共体框架计划等。一些预期在规则的刺激下,儿科研究会增加的会员国早已开始逐步建立自己的相关网站。例如,英国于2005年启动了儿童用药临床研究网络(MCRN),该网络由8个地方研究网络(LRN)组成,旨在提供卫生服务设施以支持儿科治疗、诊断和预防试验。MCRN与国家卫生系统儿童保健服务网络共同为临床随机试验和其他研究提供物质与人员支持。
儿科使用标志(Paediatric use symbol)。所有具有儿科适应症许可的产品都被要求在包装标签上设立新的标志,这可以防止对适应症许可药物的忽视。这标志是欧洲委员会在PDCO推荐的基础上挑选出来的,药品说明书上会对他的含义作出解释。其产品在标志印刷前已经获得儿科用药许可的企业,有两年时间来更改自己的包装标签。
(三)我国儿童用药管理[44,45]
目前,我国尚未颁布儿童用药相关领域的政策、法规,尚未设立儿童用药专门监管机构。儿科药物临床试验研究起步晚,整体水平低。2003年8月,国家药品食品监督管理局颁布了《药物临床试验资料管理规范》,正式将儿童纳入药物临床试验对象,2004年起全国共有23家临床试验机构获得儿科临床药物试验资格认证。最近,复旦大学附属儿科医院通过了儿童临床药物试验资格认证检查。用于药品注册而开展的儿童临床试验主要为疫苗和极少数主要用于儿童的药物,如生长激素等。针对治疗用的新药尚未开展系统和规范的儿科临床试验。
(四)国内外对比及启示
儿童用药规范制度及政策的建立,是一个漫长、循序渐进的过程。儿童用药存在的诸多问题,已被公众认同。立法建立适合我国国情的儿童用药临床研究体系和儿童药品上市后的再评价系统,保障儿童用药安全、有效是迫在眉睫的问题。同时,随着我国经济实力的不断提升、科技水平的逐步提高、法制建设的完善,建立符合我国国情的儿童用药规范的法律制度体系条件已成熟。规范儿童用药涉及范围非常广泛,不但关系到药物的研发、生产、销售、使用、监管等各个领域,而且与学校、家庭、医院等社会各层面紧密相连。发达国家和地区的经验值得借鉴。
五、保障儿童用药安全有效的政策建议
(一)完善立法,规范和激励并举
1.引入“儿科独占”,推动儿童临床试验
儿童用药品种、规格、剂型少是一个存在长久的问题,这导致了药品再分现象。虽然儿童药品的市场份额巨大,但相对成人药品市场而言,儿童药品用量不如成人大,市场空间相对较小。儿童药品的开发周期较长、研究费用高、资金投入风险大、利润较低,不少制药企业不愿意涉及。同时,儿童药品由于生产的批量小、批次多、工艺相对复杂,造成生产成本较高。在此政府有缺位的表现,缺乏立法激励与引导企业投资开发儿童专用药品。《药品管理法》中没有专门规定儿童药品的管理,在资金和专利保护等方面缺少对企业的支持;在国家中药保护品种的规定中,也没有鼓励企业开发儿童专用中药制剂的相关内容。
儿童药品临床试验的开展也有相当大的难度,虽然2003年8月国家食品药品监督管理局颁布的《药物临床试验质量管理规范(GCP)》中,正式将儿童纳入药物临床试验对象,规定在伦理与严格的监督下,儿童可以享受与成人一样的经新药临床试验后获得循证医学数据进行安全有效治疗疾病的权益,但儿童药物临床试验在我国还处于起步阶段。首先,正如我们上面所提到的,对企业来讲,进行儿童药物临床试验开发儿童专用药品或在药品说明书上增加儿童用药资料,其经济驱动力小。其次,如调研到的结果,目前国内儿童药物临床试验基地少,医务人员缺乏儿童临床试验的专业知识,相应的设备和医疗技术跟不上,一些国外企业在国内找不到合作者。另外,对于儿童药物临床试验的必要性目前看法不一,在本次调研中这一问题是存在最大争议的。本次调研的对象均为医务专业人员,在这个群体中亦有30.4%的认为在儿童群体进行临床试验是不必要、不可行的。可以推测如果调研群体再扩大些、涉及的社会群体再广些,这一比例将继续上升。这与我国居民医药知识的缺乏有关,将造成志愿者不足的现象。我国目前没有儿童药物临床研究的专门立法和行政规定,迫切需要从立法及行政规定方面给予引导。可借鉴美国和欧盟的做法,引进“儿科独占”条款,激励制药企业对儿科群体进行临床研究。
2.建立相关体系,强制儿童用药信息的完善
设立专门的职能部门,制定一系列儿童临床试验的标准文件,规范并监管儿童临床试验的各个环节是完善立法的关键。在引进激励机制的同时,应对儿童群体临床试验提出要求。新药上市申请,必须具备儿童用药的研究结果和信息。已上市药品添加新适应症、新剂型时,也应提供相关儿童用药的信息。卫生行政部门可根据现临床用药情况、ADR常见药物及类型,可建立“儿童常用药物目录”,建议或强制制药企业根据儿童临床试验,对目录内药物的儿童用药信息进行补充和修正。建立研究结果公布程序,做到所有研究分析结果广泛公布,极力避免重复试验。上海市医疗水平较高,有全国知名的儿科专科医院,可在此基础上充分利用资源,由卫生、药监部门牵头,联合儿科学组、专科医院和研究单位共建“上海地区儿童用药研究网络”平台,促进本地儿童用药的临床与研究水平。
3.规范药品说明书,加强上市后监管
我国《药品包装、标签和说明书管理规定》指出,药品说明书应包含有关药品的安全性、有效性的重要科学数据、结论和信息,用以指导安全、合理用药,这包括儿童用药,如尚不明确,应注明“尚不明确”字样。而目前儿童用药中药品说明书不规范现象还很严重。在此次调研中,有60.3%的专业人士认为儿童用药说明书不规范,位于现存问题的第2位。不规范主要表现在:儿童用药的用法用量标注不明确,与儿童有关的注意事项标注缺乏或不明确,儿童药代动力学标注较缺乏,同成分、同剂型药品的生产厂家不同、说明书标注内容有差异等,注射剂中儿童用药标注率较高,国产药品标注率低于进口和合资药品,尤以中药制剂最明显。药品说明书是药品信息的重要来源,是指导医药人员和患者合理用药的科学依据,并具有一定的法律效力,因此其内容应完整而严谨。首先,应立法对儿童用药说明书中所立项目明文规定,并区分处方药与非处方药的要求。非处方药直接向消费者销售,要求该类标签必须含有足够的用药指导,用语科学且通俗易懂,普通公众易于理解。处方药的说明书一般是供医师等专业人员参考,内容专业性强,要求全面、客观、准确,且应在外包装和说明书上增添标志警示。其次,药品说明书的改进应与儿童药品临床试验的推进和药品上市后的监管和再评价密切相连。给予儿童没有经过确认是最合适的药物,这本身就是一种不符合伦理道德的行为,这一问题只有通过临床试验才能解决。关于这一点前文已经提及,在激励企业进行儿童群体临床试验后,如可增加适应症应及时上报并体现在说明书中,药监部门在审批过程中应给予优先。某些药物的ADR只有在大规模人群中才能发现,因此药品上市后需要进行广泛的监察,而儿童特别是低龄儿童对ADR主述的能力差,特别应该加强监测。目前,我国ADR的呈报已形成了一套制度。本次调研显示88.3%的医务专业人员对ADR上报系统已有一定的认知度和熟悉度,应毫不放松加强市场监管,鼓励药厂、医疗机构积极呈报儿童ADR,根据ADR监测结果修改说明书。目前,上海市药品不良反应监测中心已有一套较为成熟的ADR上报系统,如何在此基础上进一步挖掘数据,设立儿童用药预警指标,有待进一步深入研究探讨。药品说明书中应着重描述药品的不良反应、注意事项和禁忌症,包括给药方法及给药剂量(常用治疗剂量和极量)。当然,在审批和修订过程中的严格把关是必不可少的,这是问题的关键。而对于在儿童中不能使用的药物,更应在说明书或外包装上明显标出,给予家长、医生以“警示”。
(二)加强医院管理,提高合理用药水平
医院是儿童用药的主要、关键场所,也是避免不合理用药的关键。为了保证合理用药,医、药、护、管理者、患者及其监护人都应在职责范围内为保障合理用药而协力形成体系,使完整的用药流程得以监测与完成。对临床医务工作者应该加强医德、医风教育,增强其责任心,同时医院也要严格监督管理机制,从制度上对医师的处方量进行严格的控制,坚决制止多种类合并用药等不合理用药现象,建立健全处方分析制度,定期检查医师的处方并进行分析,规范和约束医师的不合理用药行为。医疗机构要积极加强医师的培训和继续教育工作,针对儿科用药的特殊性,进行合理用药的科学研究,定期组织儿科临床药学或医学专家为医师作有关合理用药新知识、新进展的学术报告,提高儿科医师合理用药水平。临床医生应严格掌握输液治疗的适应证和使用原则,视病情及小儿用药特点尽量控制临床输液,能口服则不肌内注射,能肌内注射则不输液,防止输液滥用,同时要更加严格地控制抗菌药物的使用。
加强临床药师的培训与管理制度,加快临床药师的培养,给患儿提供专业的药学服务是避免出现儿童用药不规范的一个有效途径。药师应当熟悉儿科用药的相关专业知识,积极参与临床用药,指导临床药物的使用,建立临床药师下病房制度,对患儿家长详细讲解用药说明,保证患儿家长对用药有清楚的理解。发药同时应多宣教,可在门诊开展用药咨询工作。用药咨询服务能够改变传统的药师形象,促进药师自身业务的提高,加强与医护人员与患者的交流。改善儿科治疗药物监测,建立儿科专属治疗范围。由于药物的药动学及药效学存在年龄上的差异,靶细胞与血中药物浓度比例也存在年龄上的差异,不能用成人的参考值反映儿童的临床疗效及毒性,建立常用儿科药物的药物浓度监测系统非常重要。对儿科的常用药物,开展治疗监测,保证婴幼儿及儿童合理用药,避免受不必要的伤害。
(三)建立健全儿科基本药物目录,补充完善儿科医保目录
基本药物制度是WHO于1975年向世界各国特别是发展中国家推行的一种医疗保健制度,是政府为满足人们重点卫生保健需要和合理利用有限的医药卫生资源,保障民众用药安全、有效、合理而推行的国家药物政策,是药品监督管理的基础。我国于1979年着手基本药物工作,成立“国家基本药物遴选小组”,开始基本药物遴选。《国家基本药物目录(基层医疗卫生机构配备使用部分)》(2009版)已于2009年9月21日起施行。但目前还未建立起“儿童基本用药目录”,且在基本药物目录中,化学药部分明确标明儿童使用具体用量的只有5个品种,而中成药部分仅有一个小儿专用品种,在儿童感冒、咳嗽、健脾、消食等中成药有优势的领域都没有儿童专用药物。应当及时响应WHO的号召,并抓住我国医药卫生体制改革的机遇,建立起符合我国儿童疾病特征的《儿童基本药物目录》。另外,此次调研中专家指出,医保目录中儿童用药品种少,应加以完善。off-label用药在现阶段广泛存在且不可避免,如何规范off-label用药,避免医患矛盾值得卫生行政部门探讨。
(四)加强社区卫生服务中心儿科建设,加强正规宣教
我国现处于社会主义初级阶段,居民的文化水平较低,医药知识的缺乏是一个严重隐患;同时,我国的社区卫生服务中儿科是最薄弱的环节,很多地方还是空白,社区卫生知识普及工作也十分薄弱,尤其是在广大的城郊和农村地区,多数家长对药品说明书中药品的用法用量、适应症以及处方的理解存在着较大的误差。家庭用药是临床用药的重要组成部分,目前家长给患儿自用药物的现象普遍,包括处方药。限于家长对医药学知识、治疗方法、药品的理解程度和现有儿童用药剂型的缺乏,往往会导致用药不当。要加强社区卫生服务中心儿科的建设,定期给予儿科医生和药师专业培训,使其成为儿童保健宣教、基础疾病诊治的第一线,儿童卫生保健的基础。应加大对家长的宣传力度,利用一对一口头宣教、讲座、出版宣传墙报、发放知识手册、播放专业影碟、走出院门到社区义务宣传等多种形式的健康教育手段,帮助家长充分认识滥用药物的危害性,正确地储存、使用、管理药品。患儿家长应通过多种途径接受医药知识的普及教育和学习,改进平时家庭用药中的不正确行为,减少家庭用药不当事件的发生。
参考文献:
[1]常怡勇:《一个让人割舍不下的话题——儿童用药成人化》,《开卷有益(求医问药)》2008年第3期,第4~5页。
[2]郑映、鲍仕慧、李菁:《儿童用药的现状与思考》,《医药导报》2008年第5期,第544~545页。
[3]杨怡,张健,陆晓彤:《儿童用药安全性和有效性影响因素分析》,《中国药房》2007年第2期,第155~156页。
[4]倪韶青、寿洪初、王珏:《关于儿童用药的问题及建议》,《中国医院药学杂志》2007年第6期,第815~817页。
[5]宋新文:《儿童用药安全的三大隐患》,《中国食品药品监管》2008年第5期,第62~65页。
[6]陈伟谊、白艳甫:《儿童用药现状分析及建议》,《中国卫生经济》2005年第12期,第87~88页。
[7]王刚、谷容、刘彬:《398例儿童药物不良反应报告分析》,《儿科药学杂志》2006年第5期,第9~11页。
[8]王次霞:《儿童药物不良反应相关因素分析》,《中国医药导报》2009年第22期,第186~187页。
[9]梁雁、鲁云兰:《儿童药物不良反应及相关因素》,《儿科药学杂志》2008年第3期,第9~11页。
[10]刘瑞莉:《儿童药物使用中的不良反应》,《中国现代药物应用》2009年第4期,第148~149页。
[11]倪虹:《儿童药品不良反应80例分析》,《医药导报》2007年第5期,第508~509页。
[12]张宇靖、张昌斌:《197例儿童药品不良反应报告分析》,《药物不良反应杂志》2005年第5期,第379~381页。
[13]倪韶青、寿洪初、王珏,等:《儿童Unlicensed和Off-label用药状况调查》,《中国药学杂志》2008年第3期,第235~236页。
[14]谢奕如、蔡德、佘白蓉等:《浅析当前药品说明书存在的问题及建议》,《中国药房》2008年第4期,第311~313页。
[15]邓立新:《我院351份药品说明书中儿童用药情况分析》,《中国药房》2008年第20期,第1594~1596页。
[16]梁宇锋、邓海梅:《浅析当前药品说明书存在的问题及建议》,《齐鲁药事》2008年第10期,第656~658页。
[17]张敏:《儿童专用非处方药说明书》,《江苏药学与临床研究》2004年第5期,第63~64页。
[18]张志华、何周康、吴浩:《药品说明书中有关儿童用药内容的调查》,《儿科药学杂志》2004年第5期,第41~42页。
[19]邹慧龙、林伟萍、吴明东:《367份药品说明书中有关儿童用药情况调查》,《中国药师》2006年第3期,第277~278页。
[20]李立安、曾玲、盛应根:《525种药品说明书调查分析》,《医药导报》2007年第1期,第104~105页。
[21]张克敏、贾亦芊:《儿童用中成药说明书调查分析》,《儿科药学杂志》2008年第1期,第36~37页。
[22]苏洽玉、陈海坤:《558份化学药品说明书的调查分析》,《中国药房》2008年第4期,第316~318页。
[23]吴丽华、宋启洪、张俊鹏,等:《121种儿科常用药的药品说明书调查与分析》,《中国药业》2008年第17期,第48页。
[24]彭翠英、何周康:《507份药品说明书内容现状调查分析》,《儿科药学杂志》2009年第4期,第45~47页。
[25]蔡昭和、许江涛、蔡作忠:《189份药品说明书调查分析及改进建议》,《食品药品监管》2009年第16期,第9页。
[26]王永红、高山:《200份药品说明书评析》,《新疆医学》2009年第39期,第139~140页。
[27]Federal Food,Drug,and Cosmetic Act(FD&C Act)[EB/OL].[2009-10-08].http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm.
[28]Best Pharmaceuticals for Children Act[EB/OL].[2009-10-08].http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/Sig-nificantAmendmentstotheFDCAct/ucm148011.htm#sec2.
[29]Guidance for Industry Qualifying for Pediatric Exclusivity Under Section505Aof the Federal Food,Drug,and Cosmetic Act[EB/OL].[2009-10-08].http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM080558.pdf.
[30]胡兴、李宁、邓晓林:《美国NIH科研项目管理模式及其启示》,《泸州医学院学报》2003年第5期,第471~472页。
[31]ZAJICEK A.The National Institutes of Health and the Best Pharmaceuticals for Chil-dren Act[J].Paediatric Drugs,2009(1):45-47.
[32]Pediatric Research Equity Act of 2003[EB/OL].[2009-10-08].http://frwebgate.access.gpo.gov/cgi-bin/getdoc.cgi?dbname=108_cong_public_laws&docid=f:publ155.108[EB/OL].[2009-10-08].
[33]Retrospective review of information submitted and actions taken in response to PREA 2003[EB/OL].[2010-05-08].http://www.fda.gov/downloads/Drugs/Develop-mentApprovalProcess/DevelopmentResources/UCM197636.pdf.
[34]Guidance for Industry:how to comply with the Pediatric Research Equity Act[EB/OL].[2010-05-08].http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM077855.pdf.
[35]胡扬、赖琪、蒋学华等:《美国药品标签和说明书的法规管理》,《中国药房》2008年第7期,第490~493页。
[36]叶云、肖顺汉:《美国处方药品说明书数据结构和数据内容的解析与启示》,《中国执业药师》2009年第7期,第32~36页。
[37]杨志敏、赵德恒:《建立新药儿科临床试验管理体系的探讨》,《中国临床药理学杂志》2009年第6期,第547~549页。
[38]周鹍:《儿童药物临床试验的重要性与特殊性》,《儿科药学杂志》2009年第5期,第5~7页。
[39]Regulation(Ec)No 1901/2006of the European Parliament and of the Council of 12 December 2006on medicinal products for paediatric use and amending Regulation(EEC)No 1768/92,Directive 2001/20/EC,Directive 2001/83/EC and Regulation(EC)No 726/2004[EB/OL].[2009-10-08].http://ec.europa.eu/health/files/eudralex/vol-1/reg_2006_1901/reg_2006_1901_en.pdf.
[40]Paediatric investigation plans(PIPs),waivers and modifications[EB/OL].[2010-05-08].http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/doc-ument_listing/document_listing_000293.jsp&jsenabled=true.
[41]DUNNE J.The European regulation on medicines for paediatric user[J].Paediatric Respiratory Reviews,2007,(8):177-183.
[42]PANDOLFINI C,BONATI M.European paediatric research and children’s therapeutic needs[J].Acta paediatrica,2008,97:1232-1237.
[43]ROBERTS R,RODRIGUEZ W,MURPHY D,eta l.Pediatric drug labeling:improving the safety and efficacy of pediatric therapies[J].JAMA,2003,290(7):905-911.
[44]靳婷、高军:《〈药品说明书和标签管理规定〉中的美中不足》,《首都医药》2006年第15期,第11~12页。
[45]于培明、黄泰康:《药品说明书的法律定性必须明确》,《中国药业》2007年第4期,第14页。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。









