



第三节 用于修饰与放射性标记核酸的酶
除限制性内切核酸酶外,用于核酸操作的还有DNA聚合酶、RNA聚合酶、核酸外切酶等许多其他酶类,了解这些酶的特性与用途对从事分子生物学的工作者来说是十分必要的。厂商提供的酶制剂一般质量都非常好,只要维持酶在反应混合物中的稳定性,提供合适浓度的底物,酶就可发挥最高的活性。
一、依赖DNA的DNA聚合酶
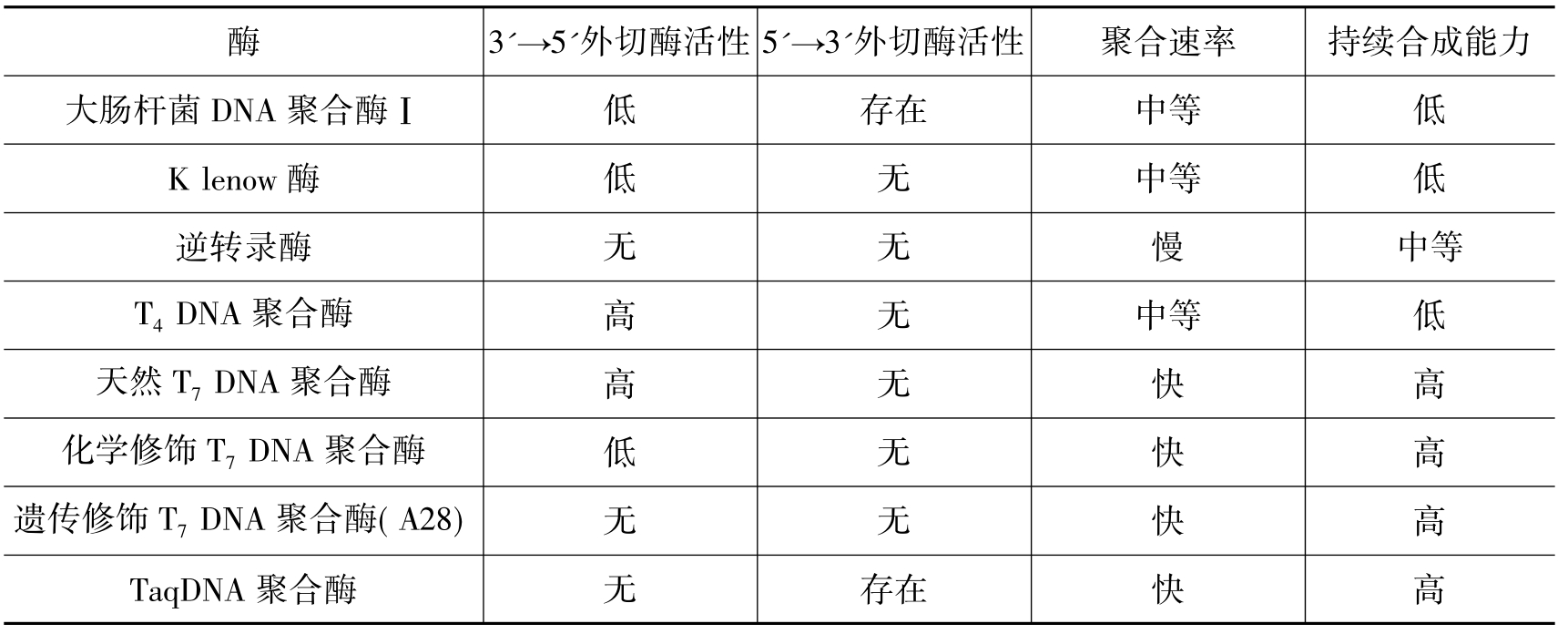
DNA聚合酶可在带引物的双链DNA分子的3'羟基端加上脱氧核苷酸,以使新生链以5'端向3'端延伸。此合成反应需要4种脱氧核苷三磷酸(dNTP),以与模板互补的方式加入。另外,DNA聚合酶发挥活性还需要镁离子的参与(见图1-7)。一些DNA聚合酶还具有3'→5'方向的外切酶活性(见图1-8),在无dNTP时,可从3'羟基端降解单链或双链DNA分子。dNTP存在时,聚合酶活性抑制其对双链DNA分子的外切酶活性。另外一些DNA聚合酶(如大肠杆菌DNA聚合酶Ⅰ、Taq DNA聚合酶)还具有5'→3'外切酶活性(见图1-9),可从5'羟基端降解双链DNA。这种外切酶活性可一次移去一个到数个核苷酸,生成5'-核苷单磷酸,或长度为10个核苷酸的寡核苷酸。各种DNA聚合酶的性质总结于表1-5。
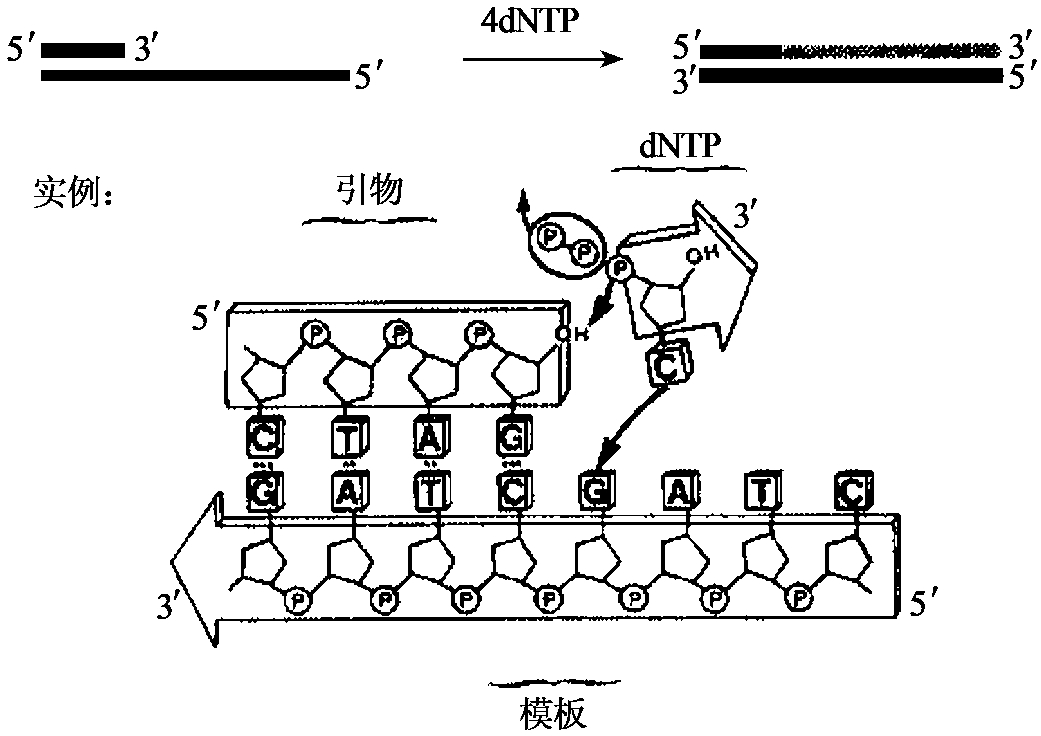
图1-7 DNA聚合酶的5'→3'聚合酶活性
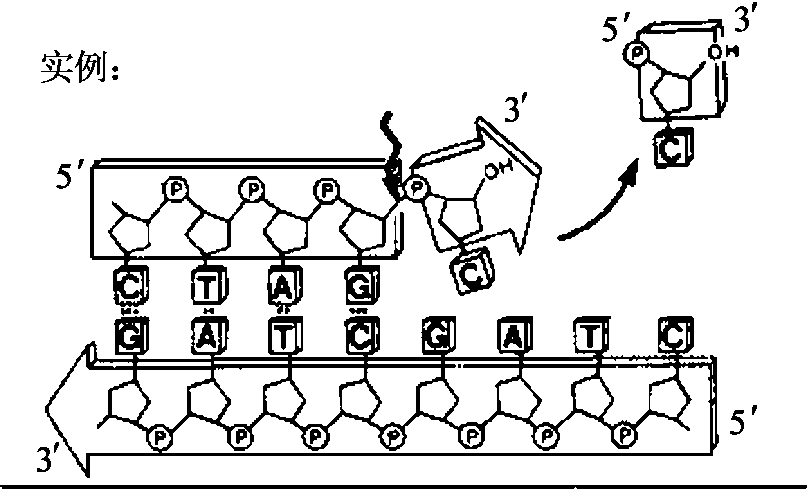
图1-8 DNA聚合酶的3'→5'外切酶活性
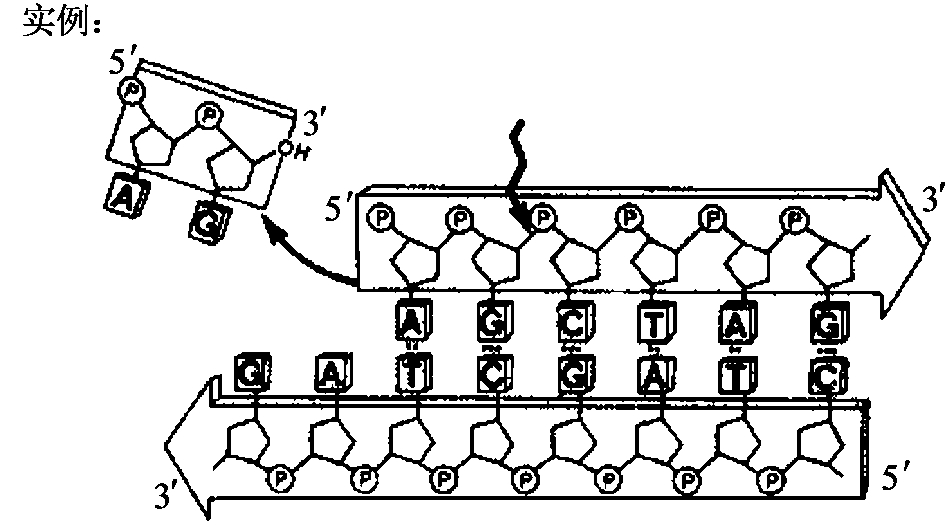
图1-9 DNA聚合酶的5'→3'外切酶活性
表1-5 DNA聚合酶的性质
下面以大肠杆菌DNA聚合酶I为例来说明依赖DNA的DNA聚合酶的特性。大肠杆菌DNA聚合酶Ⅰ最先由美国生物化学家Kornberg于1956年发现。由单条多肽链组成,分子质量为109kU。由大肠杆菌pol A基因编码,具有3种不同的酶活性,即5'→3'DNA聚合酶活性、5'→3'外切酶活性和3'→5'外切酶活性。3'→5'外切酶活性比T4或T7DNA聚合酶活性低得多。同时还具有RNA酶H的活性,RNase H活性是大肠杆菌生存所必需的,但在分子克隆中未用到此活性。在分子克隆中常用DNA聚合酶Ⅰ的5'→3'外切酶活性来制备高比活的DNA探针。
1.反应条件
25μL反应体系:
50mmol/L Tris-HCl,pH7.5
10mmol/L MgCl2
1mmol/L DTT
50μg/mL BSA
20mmol/L 4种d NTP混合液(其中一种或一种以上为α-32P标记的d NTP)
1μg DNA,3U DNA聚合酶Ⅰ
反应体积、4种d NTP的浓度、DNA的量依据具体实验而变。加入1μL0.5mol/L EDTA或75℃加热10min终止反应。切口平移反应应在15℃进行(一般为1~3h)。DNA聚合酶催化的其他反应应在20~37℃进行15~30min。
制备高比活的DNA探针,一种或几种d NTP的浓度应减少至1~2μmol/L。例如,在25μL的反应体积中,400~800Ci/mmol32P标记的dATP 25μ Ci或3000Ci/mmol32P标记的dATP 100μCi可替代20μmol dATP。为了DNA的有效合成,任何一种dNTP的浓度都不能低于1μ mol/L,这一浓度接近米氏常数。如采用高比活(3000~5000Ci/mmol)d NTP前体,反应体积应减少,或者放射性标记的dNTP应增加以维持1μmol/L dNTP的浓度。
2.用途
(1)用切口平移方法标记DNA。在所有聚合酶中只有大肠杆菌DNA聚合酶Ⅰ能用于此反应,因为它具有5'→3'外切核酸酶活性,可以在聚合酶沿DNA链推进之前,从DNA链上去除核苷酸。
(2)此全酶原来用于cDNA克隆中合成第二链,但后来已让位于逆转录酶和(或)大肠杆菌DNA聚合酶Ⅰ大片段(Klenow)。因为后二者不含5'→3'外切核酸酶活性。大肠杆菌DNA聚合酶Ⅰ的5'→3'外切核酸酶活性可以降解寡核苷酸,而后者常作为合成cDNA第二链的引物。
(3)用于对DNA分子的3'突出单链尾部进行末端标记。此反应包括两步,首先利用3'→5'外切核酸酶活性去除DNA的3'突出单链尾而产生3'凹端。然后,在高浓度的放射性标记的核苷酸前体的存在下,外切降解反应与dNTP掺入3'端的反应达到平衡。上述反应包括从凹端或平端DNA上周而复始地去除并置换3'端核苷酸,故有时称为交换反应或置换反应。如果计划用这类反应进行末端标记,T4噬菌体DNA聚合酶是首选的酶。虽然大肠杆菌DNA聚合酶Ⅰ和T4噬菌体DNA聚合酶都可进行上述反应,但后者具有更强的3'→5'外切核酸酶活性。
许多情况下,可用单一的一种缓冲液进行限制酶切割以及其后的末端标记。但用于聚合酶反应的缓冲液不适用于所有限制酶。如果DNA聚合酶缓冲液不能用于限制酶消化反应,那么就必须将限制酶反应与末端标记分两步进行。在这种情况下,先在适当的限制酶缓冲液中切割DNA,然后用酚/氯仿抽提以去除限制酶,再用乙醇沉淀DNA并重溶于TE,最后加适当体积的10×DNA聚合酶缓冲液。
二、不依赖模板的DNA聚合酶
末端脱氧核苷酸转移酶(末端转移酶)来源于小牛胸腺,分子质量为60kU,是仅存在于前淋巴细胞及分化早期的类淋巴样细胞内的一种不寻常的DNA聚合酶。该酶催化脱氧核苷酸加入到DNA的3'羟基端,伴随无机磷酸的释放(图1-10)。末端转移酶不需要模板,但需要二价阳离子。加入的核苷酸的种类决定了对阳离子的选择,若加入的核苷酸为嘧啶核苷酸,则Co2+为首选阳离子;若加入的核苷酸为嘌呤核苷酸,则Mg2+为首选阳离子。单链DNA的掺入效率最高,对于双链DNA而言,带3'突出末端的掺入效率最高,在Co2+存在下,该酶可在任意3'末端(尽管效率不一定一致)催化有限的核苷酸聚合。
1.反应条件
50μL反应体系:
100mmol/L二甲胂酸钠(Sodium Cacodylate),pH 7.0
1mmol/L CoCl 7.0
1mmol/L DTT,4pmol DNA
20μmol/L dNTP
50μg/mL BSA
10U末端转移酶
37℃温育30min,加入2μL 0.5mol/L EDTA或75℃加热10min终止反应。d NTP的选择和它的浓度依具体反应而定。在合适的条件下,末端转移酶在30min内可在每个3'末端加上大约10个左右的dCTP。
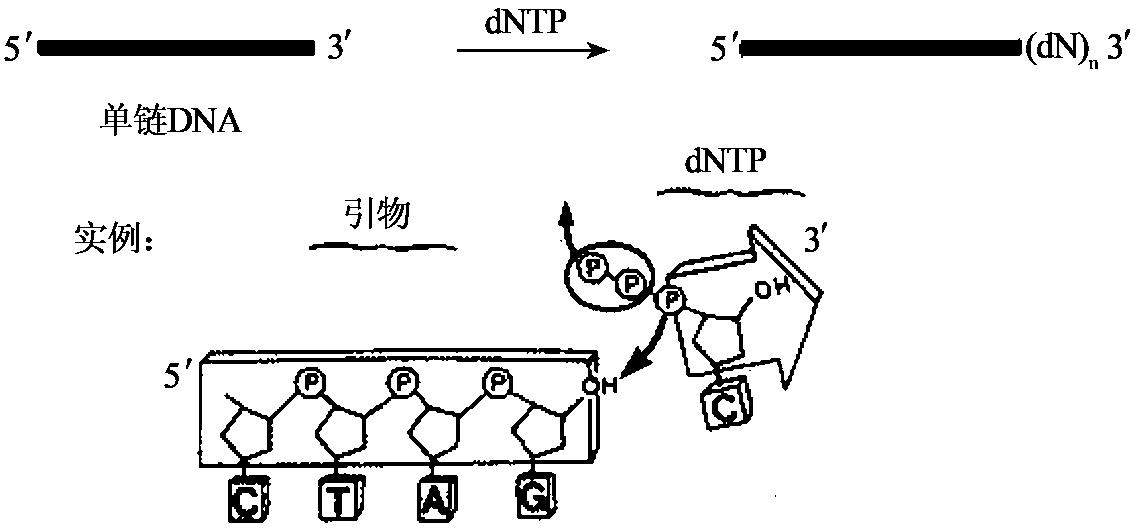
图1-10 末端转移酶活性
2.用途
(1)克隆DNA片段。在DNA片段和线性载体的末端各加上互补的同聚尾,通过同聚尾互补将DNA片段克隆至该载体。
(2)用32P标记DNA的3'末端。在DNA序列测定中,可通过32P标记的3'-脱氧腺苷三磷酸(3'-脱氧核苷三磷酸)以终止反应。
(3)在DNA片段的3'末端掺入非放射性的标签,如生物素酰化的11-脱氧尿苷,以作为荧光染料或亲和素交联物的受体位点。
(4)合成多脱氧核苷酸同聚体。
三、依赖RNA的DNA聚合酶
逆转录酶具有3种活性:①具有依赖RNA的DNA聚合酶活性,以RNA为模板,以dNTP为底物,催化合成DNA即互补DNA(cDNA);②具有依赖DNA的DNA聚合酶活性(图1-11),但是dNTP的掺入很慢,比T7DNA聚合酶的掺入慢约100倍,逆转录酶缺少对DNA的3'→5'外切酶活性;③具有从5'末端或3'末端降解RNA∶DNA杂合链中的RNA的活性,即RNase H活性(图1-12),在RNA和DNA的杂交反应中尤为有用。
现已有两种逆转录酶商品化:一种来自纯化的禽成髓细胞瘤病毒(AMV);另一种则是从一株能表达克隆化的Moloney鼠白血病病毒(MO-MLV)逆转录酶基因的大肠杆菌中分离得到的。
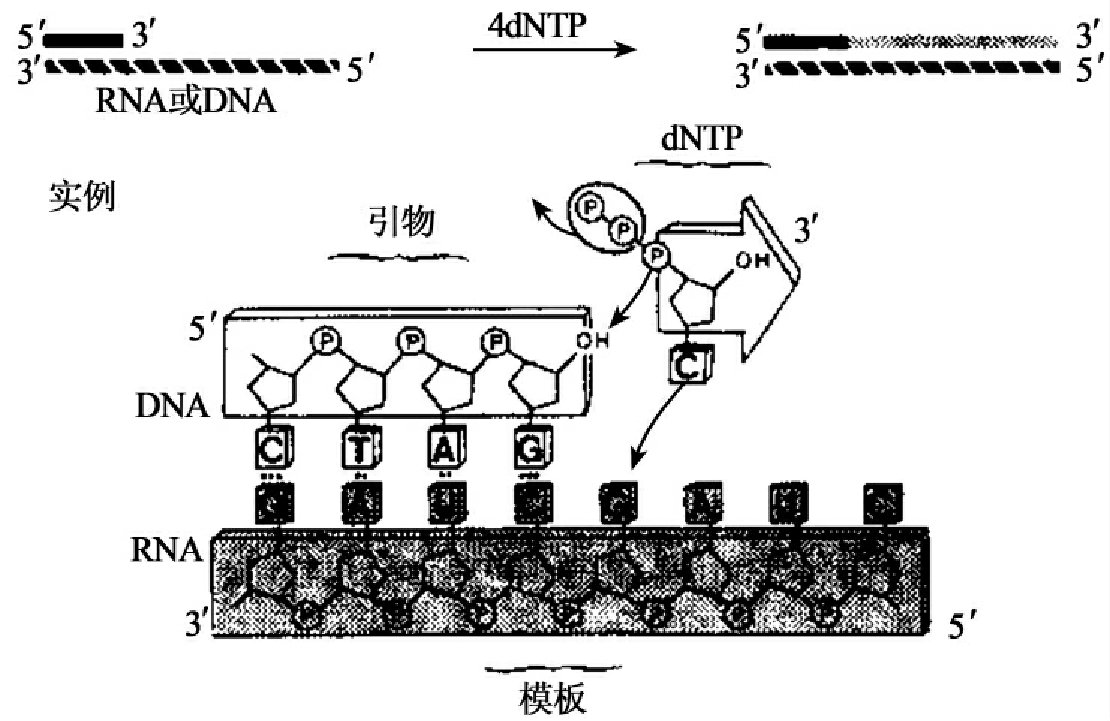
图1-11 逆转录酶的5'→3'DNA聚合酶活性
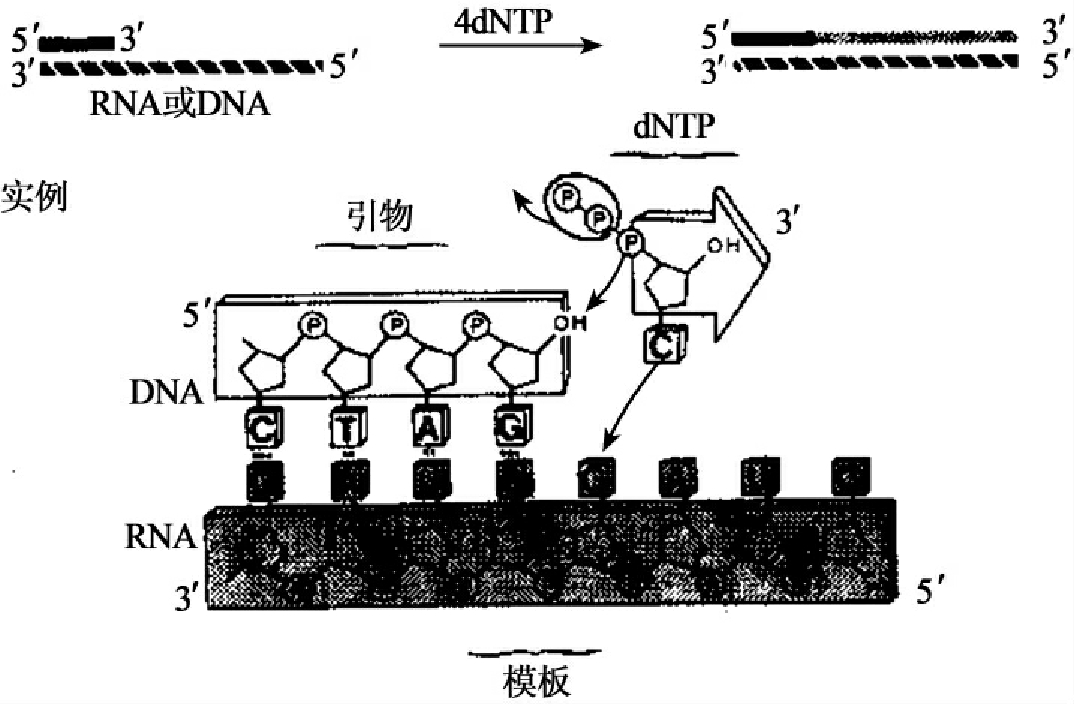
图1-12 逆转录酶的5'→3'和3'→5'外切酶活性(RNase H活性)
1.鼠源逆转录酶和禽源逆转录酶的区别
(1)禽源的逆转录酶由两条多肽链组成。它具有聚合酶活性及很强的RNase H活性。鼠源逆转录酶是单链多肽,分子质量为84kU,它具有聚合酶活性及相对较弱的RNase H活性。当合成与长段RNA链互补的DNA时,这种较弱的RNase H活性大有好处。在反应开始时,引物和mRNA模板的杂交体可成为RNase H的底物,这样cDNA合成起始时,mRNA模板的降解和合成DNA的起始竞争。另外在合成过程中如逆转录酶终止作用,RNase H即可在正在增长的DNA链的近3'端切割模板。因此,禽源逆转录酶制剂中高水平的RNase H活性往往趋向于抑制cDNA的产量并限制cDNA合成的长度。
(2)禽源逆转录酶在42℃(鸡的正常体温)能有效发挥作用。而鼠源逆转录酶在42℃则迅速失活。与鼠源逆转录酶相比,禽源逆转录酶能更有效地拷贝较复杂的mRNA。
(3)禽源逆转录酶在pH 8.3的活性要比在pH 7.6时更高,但后一种pH值更有利于鼠源逆转录酶。当反应的pH值离最佳pH值仅差0.2时,这两种酶催化合成的cDNA长度将大大降低。由于Tris的pH值随温度发生变化,因此有必要检查在所选用的温育温度下的混合物的pH值。
2.反应条件
50μL反应体系:
50mmol/L Tris-HCl pH 8.2
5mmol/L MgCl2
5mmol/L DTT
50mmol/L KCl
1μg mRNA,1μg寡聚(d T)12-18
40μmol/L 4dNTP
100μCiα-32P dNTP(比活性>400Ci/mmol)
50μg/mL BSA
40U逆转录酶
37℃温育30min。加2μL0.5mol/L EDTA或75℃加热10min终止反应。RNA模板在10μL 5mol/L NaOH中37℃温育过夜即被破坏。反应体积、4种dNTP的浓度、反应温度依具体实验而定。
3.用途
(1)逆转录酶主要用于将mRNA反转录成双链cDNA,而后者可再插入到原核载体中。在单链DNA或RNA模板参与下,也可用逆转录酶制备杂交用探针。在此反应中可用3种类型的引物:
①寡脱氧胸苷酸12~18聚体(oligo dT12-18):它结合于哺乳动物mRNA 3'端的聚腺苷酸pol(A),作为合成cDNA第一链的引物。合成时,模板3'端的序列有可能被过量拷贝,从而在cDNA中所占比例过大,这取决于逆转录酶的质量及反应条件。
②随机序列的寡核苷酸:旨在使用序列极为多样的一个寡核苷酸集群,以期至少能有一些寡核苷酸与模板退火并作为逆转录的引物。由于不同寡核苷酸结合于不同的序列上,这样在逆转录酶作用下,大部分序列都将得到拷贝,而且假如所有引物的浓度相同,则模板的所有序列得到拷贝的频率也相同。随机序列的寡核苷酸可用自动DNA合成仪合成,也可以通过水解大分子DNA获得。
③特定序列的寡核苷酸:它可作为合成与某一段特定的mRNA相对应的cDNA的引物。由于新合成的DNA与位于引物上游的mRNA序列互补,因此用这一方法(引物延伸法)可准确测出mRNA上某一固定点与其5'端之间的距离。
(2)标记带5'突出末端的DNA片段(补平反应)。
(3)当其他酶(如大肠杆菌DNA聚合酶ⅠKlenow片段-测序酶)的使用结果不理想时,逆转录酶也可用于双脱氧链终止法测序。
4.方法评注
(1)大肠杆菌DNA聚合酶Ⅰ的3'→5'外切核酸酶活性具有校对功能;但逆转录酶没有3'→5'外切核酸酶活性,因此容易出错。在高浓度dNTP和Mn2+存在下,大约每500个碱基会出现一个碱基的误掺入。
(2)逆转录酶对其dNTP的Km非常高(在毫摩尔范围内),为防止新合成的DNA的提前终止,有必要在反应体系中加入高浓度的dNTP。
(3)以寡核苷酸作引物,逆转录酶可用于合成DNA模板的单链拷贝。但是,从RNA模板合成的cDNA既可以是单链也可以是双链。以自身序列为引物进行合成时,效率远不如以外加寡核苷酸为引物时高。因此,自身互补的发夹型分子在所合成的双链cDNA中所占比例往往非常小。如有必要,在反应体系中加入终浓度为50μg/mL的放线菌素D,可同时抑制沿自身引物及外加引物合成第二链的反应。
四、依赖DNA的RNA聚合酶
用于核酸研究的有两种依赖DNA的RNA聚合酶:一种为大肠杆菌RNA聚合酶,这是一种由多个亚基组成的酶,它可识别启动子的-10区和-35区,该酶最初用于体外转录合成,但转录过早终止,因而很难得到>500个碱基的转录产物,且特异性较低,常从其他序列(如模板5'端)起始转录;另一种是噬菌体RNA聚合酶,包括SP6、T7和T3RNA聚合酶。
噬菌体RNA聚合酶与大肠杆菌RNA聚合酶相比具有如下优点:①持续合成能力很强,可催化合成数千个碱基的转录产物;②转录既快速又高效,以2μg DNA为模板,30min可转录60μg的转录产物;③对相应的启动子具有高度特异性;④T7和T3RNA聚合酶可通过基因工程方式制备,因而可以得到既价廉、特异活性又高的RNA聚合酶。
(一)大肠杆菌RNA聚合酶
大肠杆菌RNA聚合酶由α、2β、β、'σ5个亚单位组成,分子质量约450kU。它以天然的或变性的DNA为模板,以NTP为底物,催化转录合成RNA。优先从具有-10区和-35区保守序列的启动子起始转录,终止于终止子序列。转录的特异性和长度取决于DNA的纯度、启动子和终止子的长度、一价阳离子和二价阳离子的种类和浓度。缺少σ因子的核心酶不能识别启动子,因此在模板上随机起始转录。
1.反应条件
50μL反应体系:
40mmol/L Tris-HCl pH 8.0
10mmol/L MgCl2+5mmol/L DTT
50mmol/L KCl,2μg双链DNA(含启动子)
300μmol/L4NTP
50μg/mL,BSA
10UE.coli RNA聚合酶
37℃温育30min,加2μL 0.5mol/L EDTA或75℃加热10min终止反应。反应体积、DNA浓度和4种NTP含量依具体实验而变。
2.用途
(1)当DNA不能被克隆到带噬菌体RNA聚合酶启动子的载体时,可用大肠杆菌RNA聚合酶催化转录。
(2)大肠杆菌RNA聚合酶全酶可用于测定含大肠杆菌表达启动子序列的克隆DNA。
(3)缺少σ因子的核心酶在高浓度随机引物和低浓度NTP存在时,以DNA为模板合成短的相对一致的转录物。以天然DNA为模板比变性DNA为模板合成的转录物要长。
(二)噬菌体RNA聚合酶:SP6、T7、T3
SP6噬菌体可合成依赖于DNA的RNA聚合酶,该酶识别双链DNA模板上相应的噬菌体特异性的启动子,并沿此模板起始RNA的合成。利用该酶可以在体外合成大量与外源DNA一条链互补的RNA。该外源DNA克隆到专门设计的载体中,并且位于启动子下游。已有一些专用的载体可通过改变外源基因上游的启动子的方向而合成与模板任意一条链互补的RNA。
T7和T3RNA聚合酶由各自噬菌体基因1编码,能识别双链DNA模板上特异性启动子,并沿模板起始RNA合成。T7和T3RNA聚合酶在体外的应用与SP6RNA聚合酶完全一样。T7和T3RNA聚合酶已在大肠杆菌中克隆和表达。T7RNA聚合酶也已在酵母中得到克隆和表达。这样,带有T7噬菌体启动子的载体就可用于在体内表达克隆化基因。
3种噬菌体RNA聚合酶均为单一多肽链,分子质量90~100kU。每一种酶对相应的启动子有高度特异性。转录速度非常快(体外为大肠杆菌RNA聚合酶的10倍),持续合成能力很强。
1.反应条件
50μL反应体系:
40mmol/L Tris-HCl pH7.5
10mmol/L MgCl2+
5mmol/L DTT
2μg DNA模板(含相应噬菌体启动子)
400μmol/L 4NTP
50μ g/mL BSA
1mmol/L亚精胺(仅用于SP6催化的反应混合液中)
10U RNA聚合酶(T7、T3或SP 6)
37℃温育30min。加入2μL 0.5mol/L EDTA或75℃加热10min终止反应。反应体积、DNA浓度、4种NTP含量依具体反应而变。上述反应条件可催化合成60μg以上的转录产物。如要制备高比活的标记转录物,需将标记的NTP浓度降至10μmol/L。亚精胺有助于SP6RNA聚合酶的催化反应,应在室温加入到反应混合液中,以免DNA发生沉淀。
2.用途
T7、T3和SP6噬菌体RNA聚合酶可以高效、特异地转录置于相应的启动子下游的DNA序列。
(1)噬菌体RNA聚合酶可用于产生均匀标记的高比活单链RNA探针,用于DNA或RNA序列的同源性分析。
(2)均匀标记的RNA转录物可用于分析RNA或DNA的末端,用于基因组DNA序列测定和研究前体RNA的剪接。
(3)RNA转录物可用于体外翻译,在放射性标记的氨基酸存在下,合成放射性纯蛋白质。
(4)T7RNA聚合酶可用于体内高效表达克隆化基因(在T7RNA聚合酶启动子的严格控制下)。
五、不依赖DNA的RNA聚合酶
poly(A)聚合酶以ATP为前体分子,催化AMP掺入到RNA游离的3'-羟基端(图1-13)。
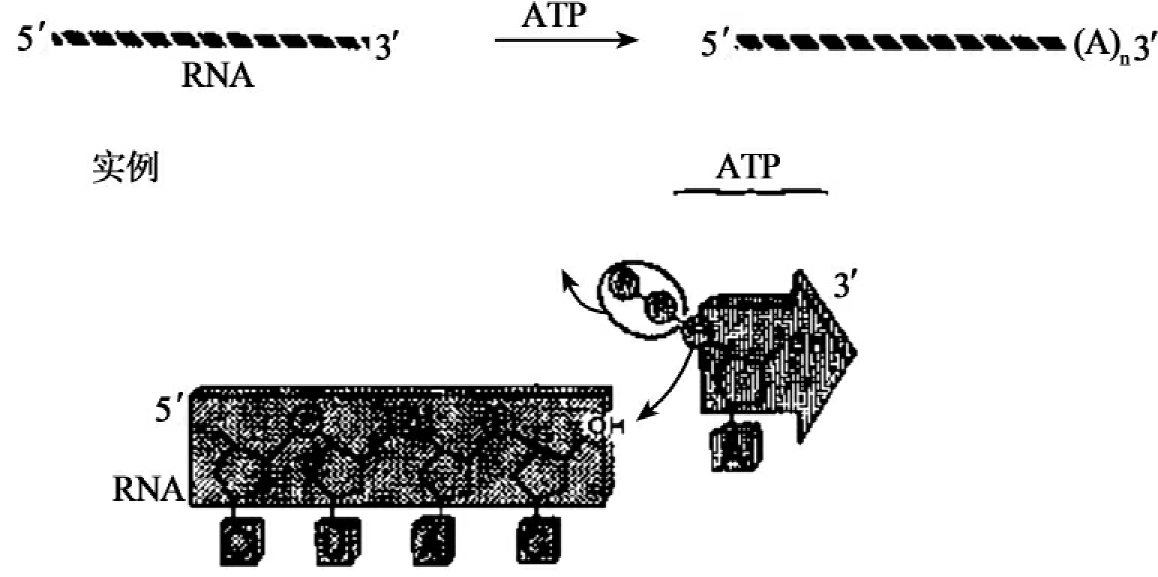
图1-13 poly(A)聚合酶活性
1.反应条件
50μL反应体系:
40mmol/L Tris-HCl pH 8.0
10mmol/L MgCl2+
2.5mmol/L MnCl2+
250mmo1/L NaCl
250μg/mL RNA
250μmol/L ATP
50μg/mL BSA
5U poly(A)聚合酶
2.用途
(1)用α-32P ATP标记RNA的3'末端。用此法标记的RNA可作为杂交探针。但对于克隆化基因,用噬菌体RNA聚合酶标记的RNA探针更有效且比活高得多。用逆转录酶合成的cDNA是标记细胞RNA的首选方法。
(2)克隆缺少poly(A)尾的RNA。用poly(A)聚合酶加尾,oligo(dT)作为引物合成c DNA。
六、磷酸酶和激酶
(一)碱性磷酸酶
细菌碱性磷酸酶(BAP)和小牛肠碱性磷酸酶(CIP)都能催化水解DNA、RNA、d NTP和NTP上的5'磷酸残基(图1-14)。这种具有5'-羟基端的脱磷酸化产物可在多核苷酸激酶催化下,用γ-32P ATP进行标记(图1-15)。两种磷酸酶的活性都依赖Zn2+,在大多数情况下,CIP更常用。因为较BAP而言,它可在70℃加热10min灭活,或通过酚抽提而灭活,此外,CIP的活性比BAP高10~20倍。
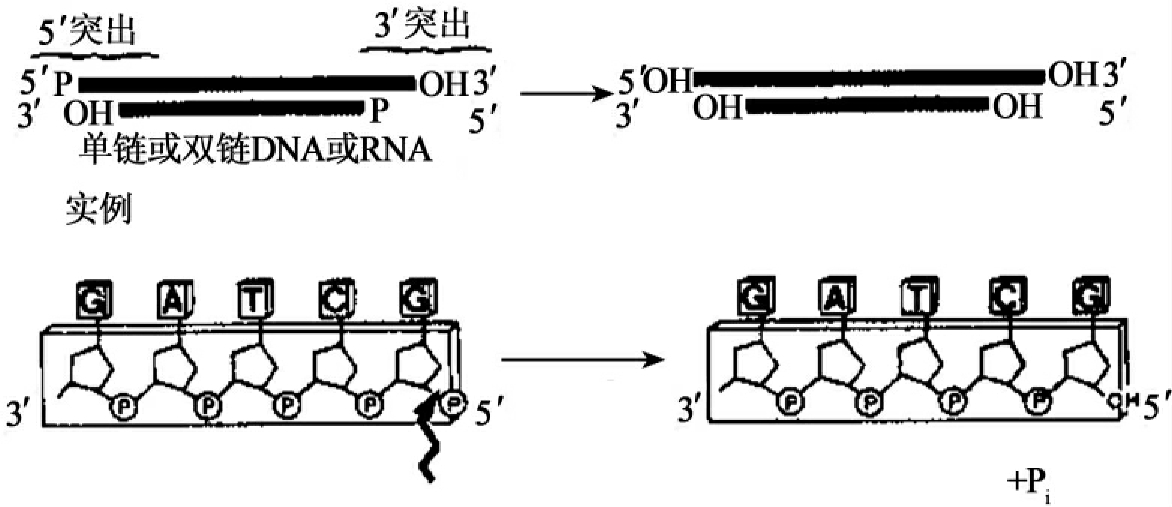
图1-14 碱性磷酸酶催化的脱磷酸反应
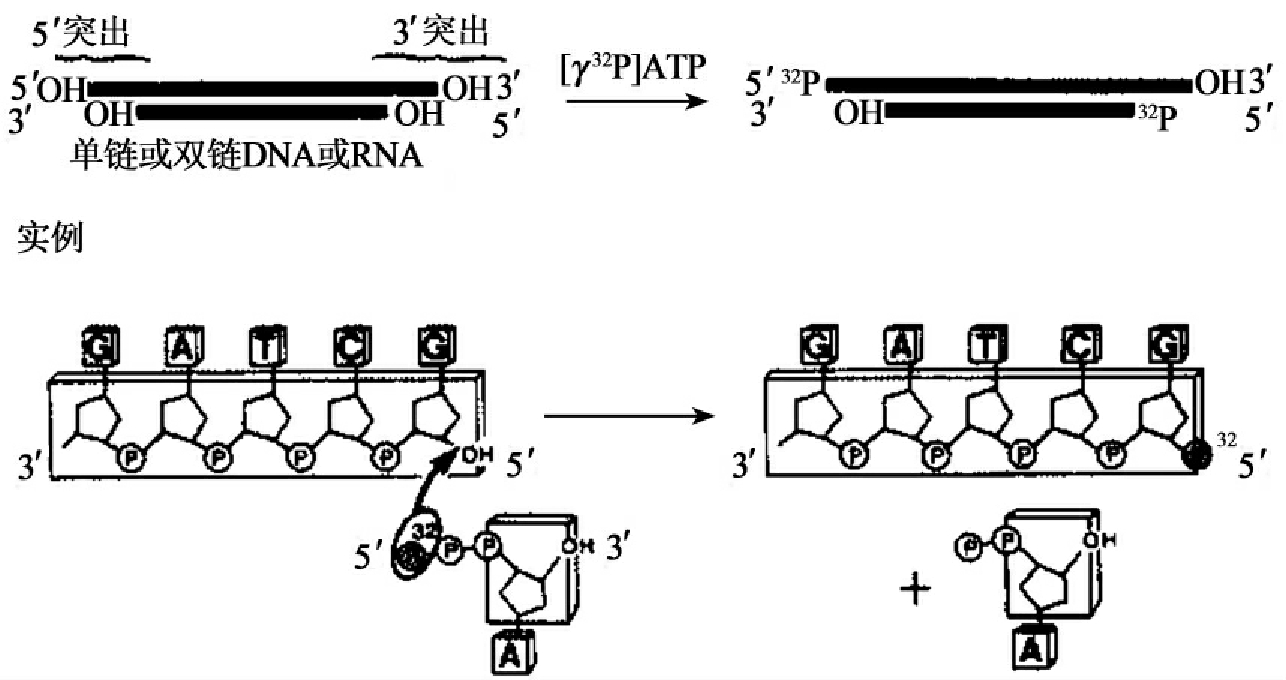
图1-15 T4多核苷酸激酶催化的磷酸化反应
1.细菌碱性磷酸酶
50μL反应体系:
50mmo1/L Tris-HCl pH8.0
1mmol/L ZnCl2
1~20pmol DNA末端
0.1U BAP
60℃温育30min。加入0.1%SDS和100μg/mL蛋白酶K,于37℃温育30min终止反应。用酚提抽2次,然后用乙醇沉淀DNA。
2.小牛肠碱性磷酸酶
50μL反应体系:
20mmo1/L Tris-HCl pH8.0
1mmol/L MgCl2
1mmol/L ZnCl2
1~20pmol DNA末端
0.1U CIP
37℃温育30min。75℃加热10min或用酚提抽终止反应,然后用乙醇沉淀DNA。
3.用途
(1)在用γ-32P ATP和T噬菌体多核苷酸激酶标记前,用碱性磷酸酶脱去5'末端的磷4酸基团,5'末端32P标记的DNA可应用于Maxam-Gilbert化学测序、RNA测序和特异性DNA或RNA片段的图谱构建。
(2)载体DNA 5'末端的去磷酸化可防止载体的自连。
(二)T4多核苷酸激酶
T4多核苷酸激酶来源于T4噬菌体感染的大肠杆菌,现已通过基因工程的方式生产。T4多核苷酸激酶具有两种活性。正向反应活性是高效率的,它可催化ATP的γ位磷酸转移至DNA或RNA的5'端羟基(图1-15),是标记或磷酸化5'末端的首选方法。交换反应活性很低,它催化5'端磷酸的交换,在过剩的ADP存在下,T4多核苷酸激酶催化DNA的5'端磷酸转移给ADP,然后DNA从γ-32P ATP中获得放射性标记的γ-磷酸而重新磷酸化(图1-16)。除磷酸化活性外,T4多核苷酸激酶还具有3'磷酸酶活性。T4多核苷酸激酶的特性见表1-6。
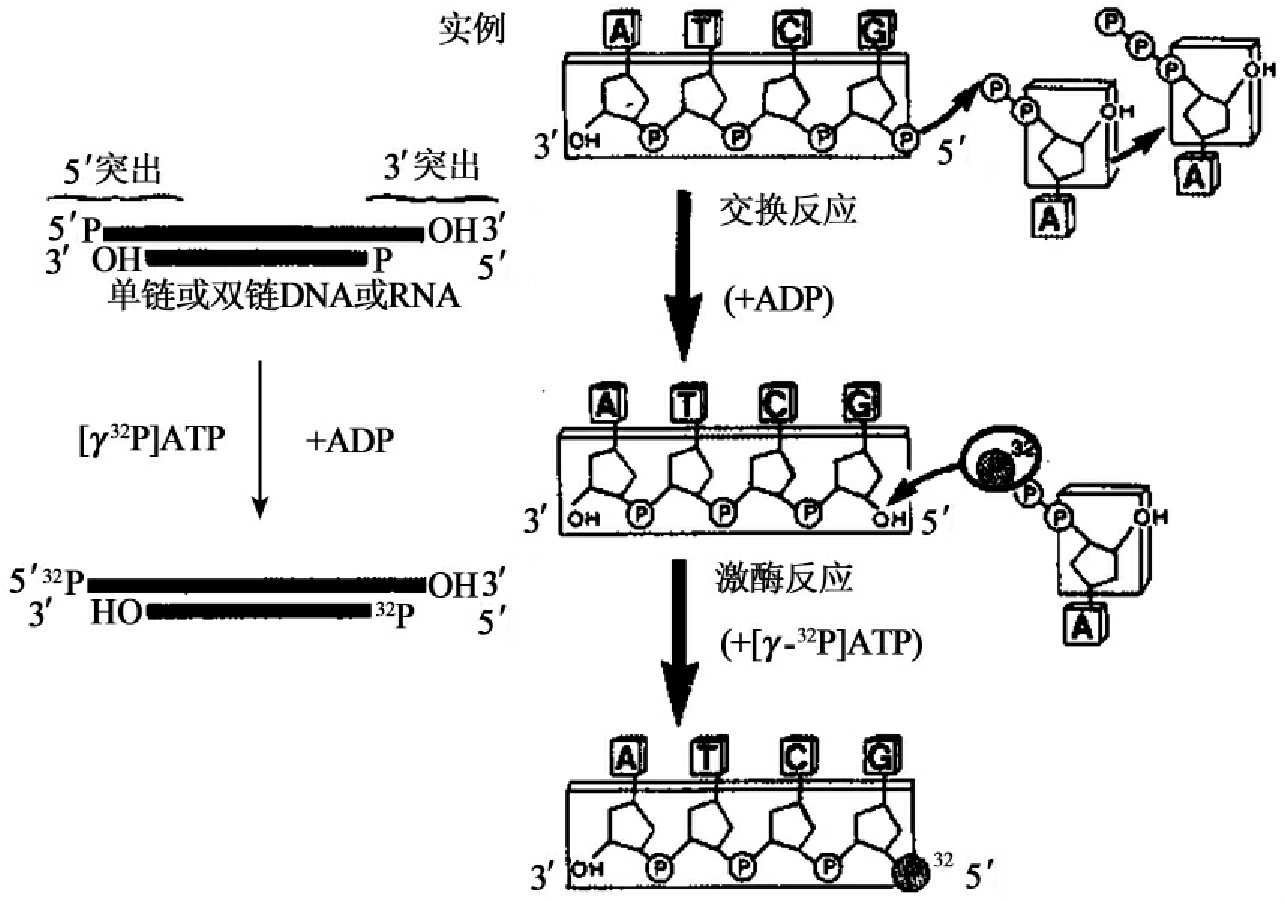
图1-16 T4多核苷酸激酶催化的交换反应
其用途如下:
(1)用化学降解法测DNA序列。
(2)通过DNaseⅠ足迹法或检查DNA免受化学物质损伤(如二甲基亚砜、甲锭丙基EDTA)的情况而确定特异的蛋白质和DNA相互作用。
(3)通过部分消化5'末端标记的DNA片段构建物理图谱;RNA转录物末端分析和DNA中内含子的位置确定。
(4)合成用于分析DNA和RNA连接酶活性的底物。
(5)标记寡核苷酸以利电泳纯化。
(6)将寡核苷酸连接至DNA载体上。化学合成的寡核苷酸具有5'羟基末端,要进行克隆必须先将它们磷酸化。
表1-6 T4多核苷酸激酶的特性
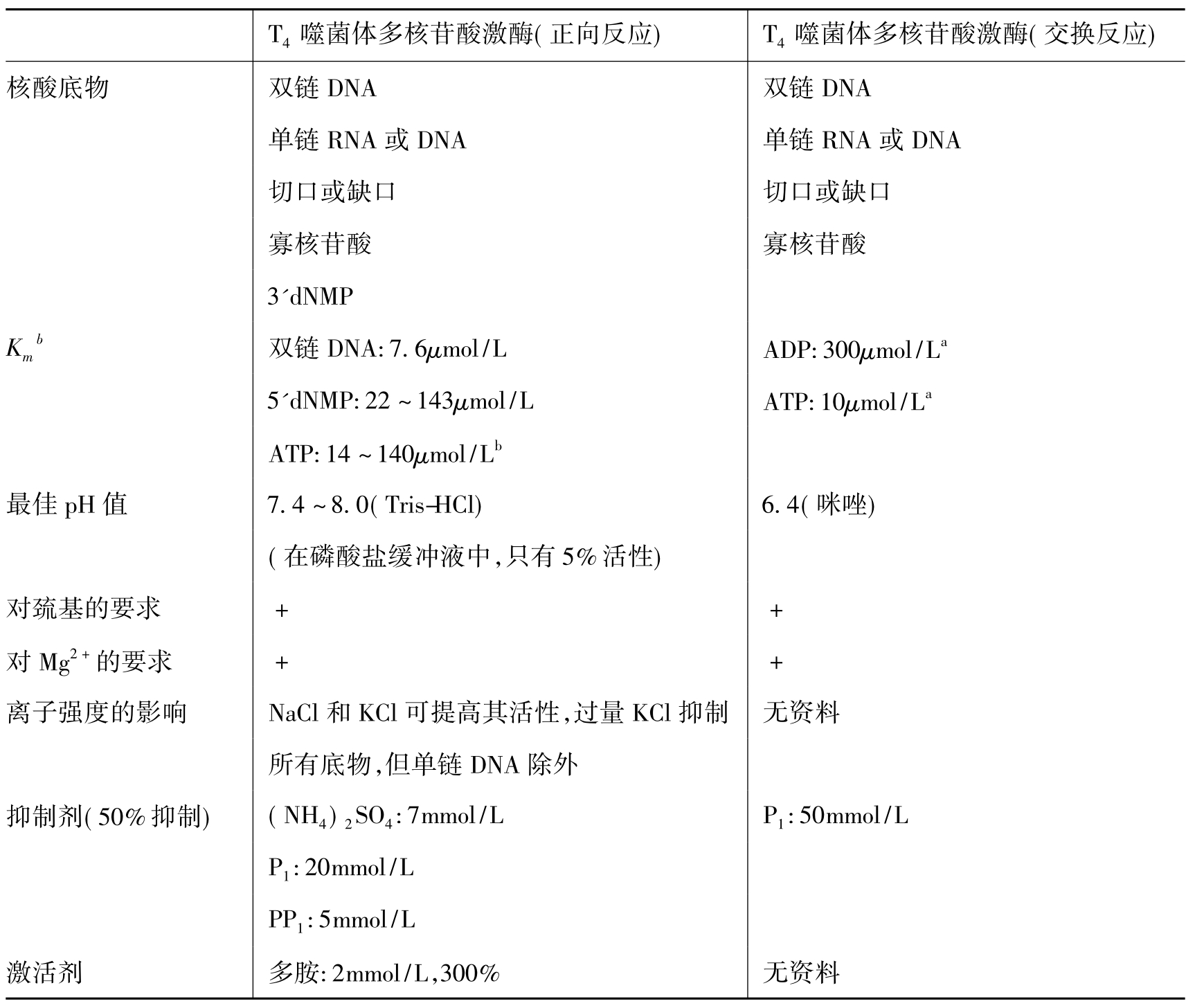
a.这是pH6.4时能使酶活性达最高的底物浓度,而并不是Km。
b.对ATP的Km因底物而不同。
七、外切核酸酶
(一)单链5'→3'和3'→5'外切核酸酶
外切核酸酶Ⅶ(exoⅦ)来自大肠杆菌的外切核酸酶Ⅶ,由两个亚基组成,由xse A和xse B基因编码。它是作用于单链DNA 3'端和5'端的外切核酸酶(图1-17),其降解产物为小的寡核苷酸,该酶的活性不需要Mg2+,在10mmol/L EDTA存在时仍保留完全的活性。
1.反应条件
50μL反应体系:
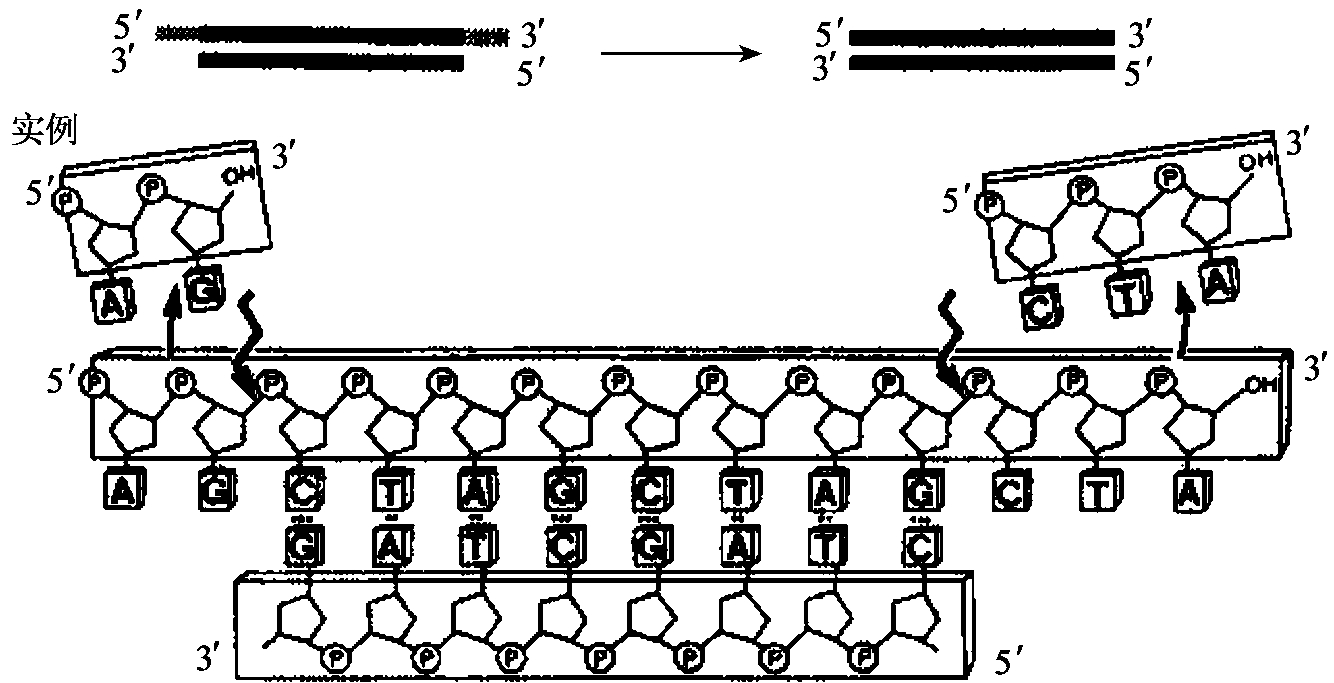
图1-17 外切核酸酶Ⅶ活性(催化DNA的降解反应)
70mmol/L Tris-HCl pH 8.0
8mmol/L EDTA
10mmol/Lβ-巯基乙醇
1pg DNA,5pg/mL BSA
20U外切核酸酶Ⅶ
37℃温育30min。酚提抽和乙醇沉淀终止反应。外切核酸酶Ⅶ活性不被EDTA抑制。
2.用途
(1)基因组DNA中内含子的位置作图。
(2)切除通过poly(dA-dT)加尾插入到质粒载体中的DNA片段。
(二)双链5'→3'外切核酸酶
1.λ噬菌体外切核酸酶(λexo)
该酶来自λ噬菌体感染的大肠杆菌,它催化从双链DNA5'端带磷酸基团的末端开始的持续水解,释放5'核苷单磷酸(图1-18)。但不降解5'端为羟基的末端。
(1)反应条件
50μL反应体系:
67mmol/L甘氨酸-KOH pH9.4
2.5mmol/L,MgCl2
2μg DNA
50μg/mL BSA
10Uλ噬菌体外切核酸酶
37℃温育1~30min,具体温育时间依消化程度而定。加入2μL 0.5mol/L EDTA,或75℃加热10min终止反应。
(2)用途
①在应用双脱氧法测序时,将双链DNA转变为单链DNA。
②去掉双链DNA的5'突出末端,便于末端转移酶加尾。
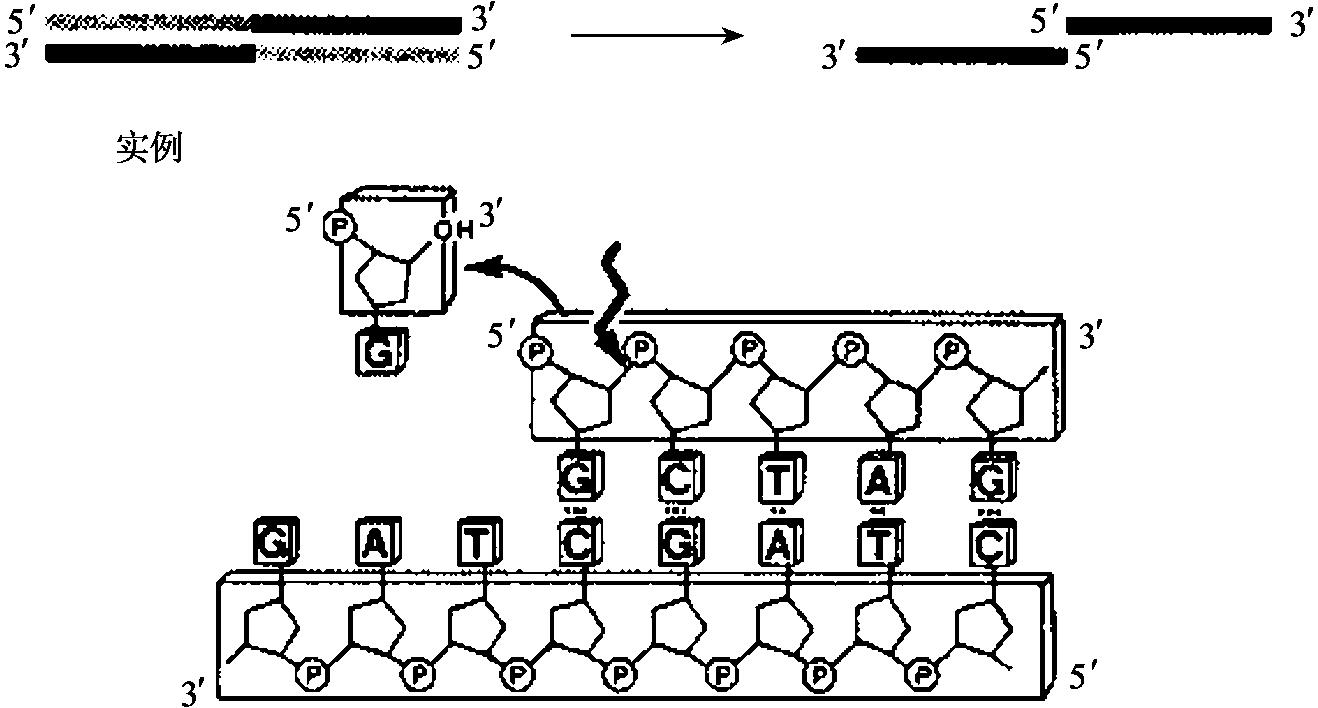
图1-18 5'→3'外切核酸酶活性
2.T7基因6外切核酸酶
该酶由T7噬菌体基因6编码,现已通过基因工程克隆表达及纯化。该酶催化双链DNA的5'末端的依次水解,生成5'核苷单磷酸。它的作用持续性较低,对5'端为磷酸基团或羟基的DNA链都能起作用。
(1)反应条件
50μL反应体系:
50mmol/L Tris-HCl pH7.5
5mmol/L MgCl2
5mmol/L DTT
20mmol/L KCl
2μg DNA
50μg/mL BSA
5UT7基因6外切核酸酶
37℃温育1~30min,具体温育时间依消化程度而定。加入2μL 0.5mo1/L EDTA,或75℃加热10min终止反应。
(2)用途
与λ噬菌体外切核酸酶相比,该酶的优势在于它从5'末端的降解均匀且可控制。此外,它也可降解5'端为羟基的末端。
(三)双链3'→5'外切核酸酶
外切核酸酶Ⅲ来自大肠杆菌,由xth A基因编码。该酶可特异性催化双链DNA从3'羟基末端开始水解,释放5'单核苷酸(图1-19)。它的外切酶活性是非持续的,因而是用于在双链DNA中产生均匀的单链区的理想工具酶。水解速率受核苷酸组成影响(C>>A-T>>G)。
此外该酶还有以下3种活性:对无嘌呤或无嘧啶位点DNA特异的内切核酸酶活性,RNase H活性及3'磷酸酶活性。3'磷酸酶活性可去除3'末端磷酸,但并不切割核酸内的磷酸二酯键。外切核酸酶活性并不降解单链DNA及带3'突出末端的双链DNA。
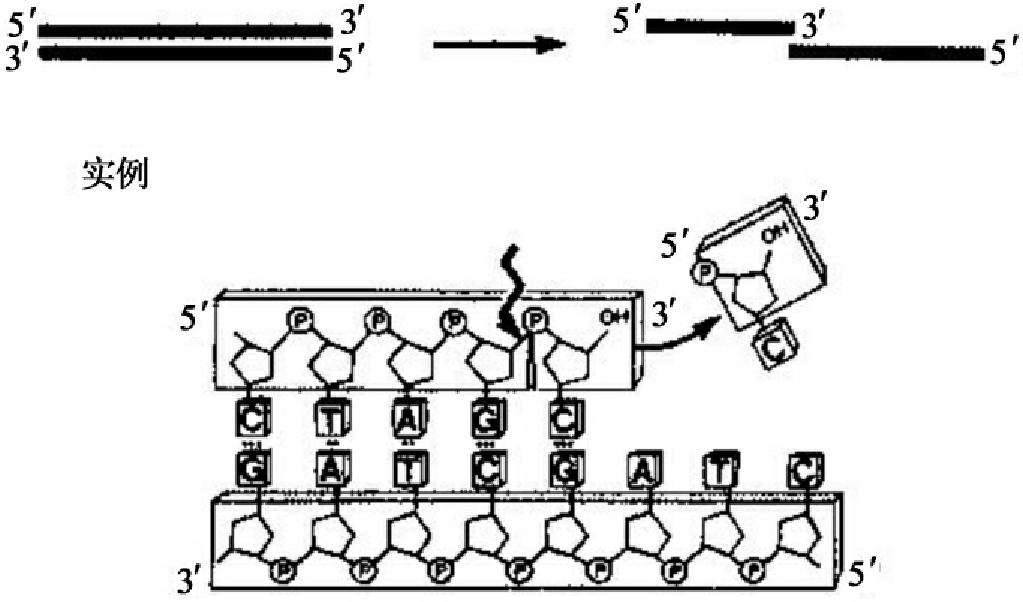
图1-19 外切核酸酶Ⅲ活性
1.反应条件
50μL反应体系:
50mmol/L Tris-HCl,pH7.5
5mmol/L MgCl2
5mmol/L DTT
2μg DNA
50μg/mL BSA,10U外切核酸酶Ⅲ
37℃温育1~30min,具体温育时间依消化程度而定。加入2μL 0.5mo1/L EDTA,或75℃加热10min终止反应。1U的为外切核酸酶Ⅲ37℃、10min内从1μg 5000bp长的线性双链DNA的每个3'凹端切除约200个核苷酸的酶量。
2.用途
(1)与Klenow酶联用,制备链特异性的放射性探针,类似于利用T4DNA聚合酶活性进行交换合成反应。
(2)制备单链模板用于双脱氧测序。
(3)从克隆化DNA片段的特定位置进行非定向缺失,构建的亚克隆可不经限制酶定位直接用于DNA序列测定。
八、内切核酸酶
(一)Bal 3Ⅰ核酸酶
Bal 3I既具有单链特异性的DNA内切酶活性,又具有外切核酸酶活性。利用其外切酶活性连续去除线状双链DNA分子3'端的单核苷酸后形成的单链DNA可被Bal 3Ⅰ的内切酶活性所降解。对于双链环状DNA,它可特异性地降解双链DNA缺口或DNA超螺旋上产生的瞬时单链区域。对于双链线性DNA,它可从DNA的3'端和5'端有节制地降解产生缩短的DNA分子。该酶亦可作为核酸酶水解rRNA和tRNA(图1-20)。
Bal 3Ⅰ的活性依赖Ca2+和Mg2+的存在。因此可在反应的不同阶段加入螯合剂EDTA而使反应终止。该酶可在十二烷基磺酸钠(SDS)和尿素中保持活性。
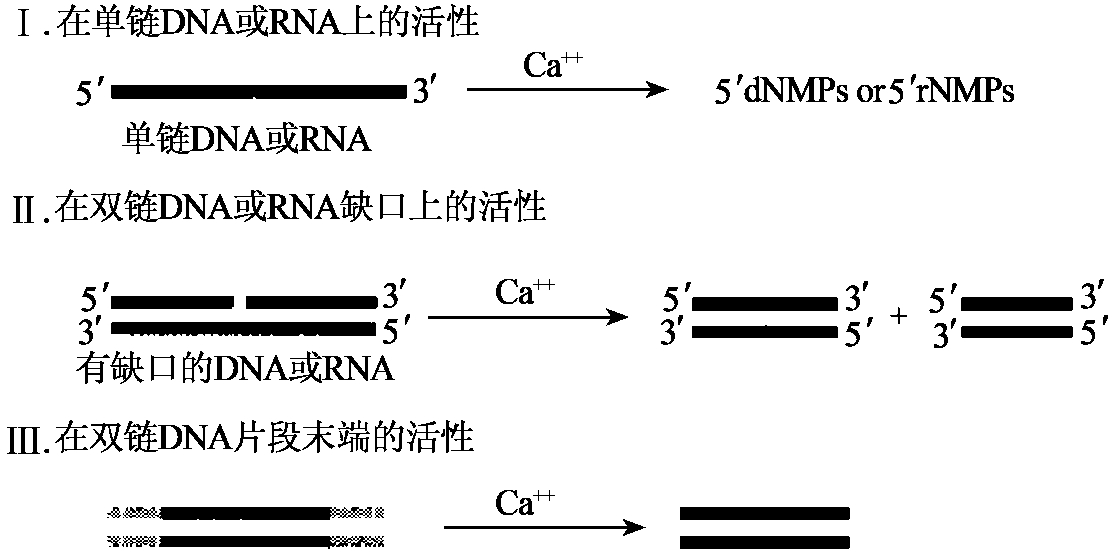
图1-20 Bal 3Ⅰ核酸酶活性
1.反应条件
50μL反应体系:
50mmol/L Tris-HCl,pH7.5
10mmol/L CaCl2
10mmol/L MgCl2
600mmol/L NaCl
2μg DNA
50μg/mL BSA
10U Bal 3Ⅰ核酸酶。
30℃温育1~30min,温育时间依消化程度而定。加入5μL 0.5mo1/L EDTA,或75℃加热10min终止反应。用琼脂糖凝胶电泳分析降解产物,用乙醇沉淀去掉NaCl便于后续的酶促反应。
1U的Bal 3Ⅰ核酸酶在30℃10min内,在50μL反应体积和50μg/mL的DNA浓度下,从线性pBR322的两端降解200个碱基对。
2.重要参数
(1)Bal 3Ⅰ可因核酸酶的活性受RNA污染物的抑制,所用DNA必须经氯化铯超离心纯化或凝胶纯化。
(2)Bal 3Ⅰ核酸酶的单位数仅供参考,对于具体的DNA模板,消化的程度应以琼脂糖凝胶电泳监测。
(3)Bal 3Ⅰ核酸酶活性受DNA组成的影响,AT丰富的区域比GC丰富区降解得更快。
(4)经Bal 3Ⅰ核酸酶消化的DNA片段可以直接用于连接,但效率较低。消化的DNA经酚抽提和乙醇沉淀后可提高连接效率。消化后DNA末端需用Klenow酶或T4DNA聚合酶补平后,再用T4DNA连接酶连接。
3.用途
(1)在可控条件下产生并克隆不同大小的缺失体。
(2)DNA片段的限制性位点作图。
(3)研究经诱变剂处理的超螺旋DNA螺旋区的变化和超螺旋DNA的二级结构。
(二)S1核酸酶
S1核酸酶是高度特异性的单链内切酶。它降解单链DNA或RNA,产生带5'磷酸基团的单核苷酸或寡核苷酸。该酶需要Zn2+,最适pH4.6。双链DNA、双链RNA及DNA∶RNA杂交体都对该酶有抗性。水解单链DNA的效率比双链DNA快75000倍。但在酶的量非常大时,双链核酸可被完全消化,中等量的S1核酸酶可在切口或小缺口处切割双链核酸(图1-21)。S1核酸酶在尿素、SDS和甲酰胺中稳定。
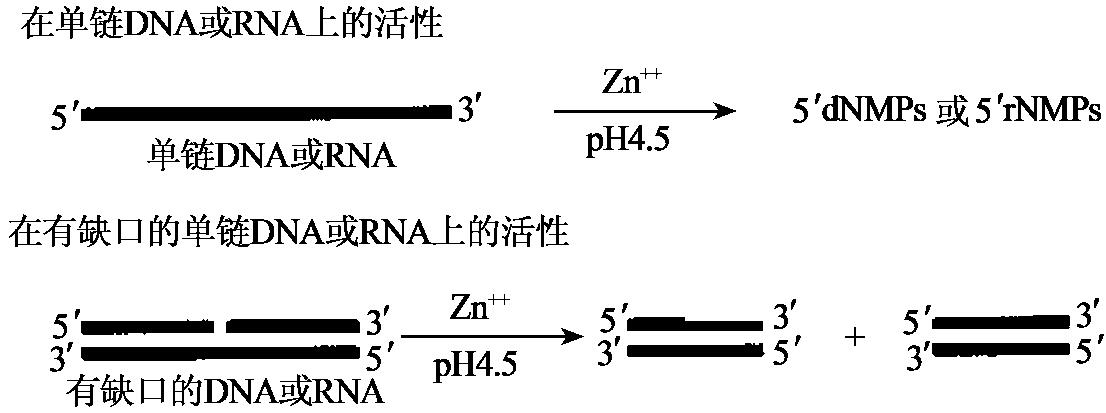
图1-21 S1核酸酶活性
1.反应条件
100μL反应体系:
50mmol/乙酸钠,pH4.6
1mmol/L乙酸锌
250mmo1/L NaCl
2μg DNA
50μg/mL BSA
10U S1核酸酶
37℃温育30min,加入1μL 0.5mol/L EDTA终止反应。反应体积、DNA浓度、酶量、反应温度和时间依具体反应而变。
2.用途
(1)分析抗S1核酸酶降解的RNA∶DNA杂合体以定位RNA转录物的5'端和3'端。
(2)通过消化成熟mRNA与32P标记的基因组DNA杂交形成的杂合体,确定内含子的位置。S1核酸酶作用于杂合分子中由内含子产生的单链环。
(3)消化在以逆转录酶合成cDNA时形成的发夹结构。
(4)去掉DNA片段的单链突出末端,产生用于连接的平端。
(5)在限制酶位点处产生小的缺失。
(三)绿豆核酸酶
绿豆核酸酶(来源于绿豆芽)是与S1核酸酶类似的高度特异性单链内切酶,最适pH5.0。当NaCl的浓度>200mmol/L时,其活性明显降低。
1U酶定义为使用单链鲑鱼精子DNA作为底物,在37℃1min内产生1μg酸溶性物质的酶的量。
1.反应条件
100μL反应体系:
30mmol/L乙酸钠,pH5.0
50mmol/L NaCl
1mmol/L乙酸锇
5%(V/V)甘油
1μg DNA
50μg/mL BSA
15U绿豆核酸酶
37℃温育30min,加入1μL 0.5mol/L EDTA。反应体积、DNA的量、酶的量、反应温度和反应时间依具体反应而变。
2.用途
(1)在转录物定位作图实验中,在紧邻最后一个杂交配对碱基处起作用,而不移去任何配对的核苷酸。
(2)准确切除经限制性内切核酸酶消化的不配对的突出端。
(四)微球菌核酸酶
微球菌核酸酶来源于金黄色葡萄球菌,是一种非特异性的核酸酶。它能切割单链和双链DNA和RNA,生成带3'磷酸基团的寡核苷酸和单核苷酸。该酶对单链核酸作用更强,它优先在DNA或RNA的AT或AU丰富区切割,其活性可被Ca2+的特异螯合剂EDTA灭活。
1U酶定义为在37℃和pH8.0时,每分钟催化天然DNA产生1μmol酸溶性寡核苷酸的酶的量。
1.反应条件
典型的消化反应条件是10mmol/L Tris-HCl,pH8.0,1mmol/L CaCl2。但在高pH值条件下,活性更高。反应体积、核酸的来源与量、酶的量、离子强度、反应温度与时间依具体反应而有很大的不同。反应可被EDTA或EGTA终止。
2.用途
(1)研究染色体结构。
(2)从未破坏酶活性的破细胞粗提物中去除核酸。
(3)消化在以逆转录酶合成cDNA时形成的发夹结构。
(4)去掉DNA片段的单链突出末端,产生用于连接的平端。
(5)在限制酶位点处产生小的缺失。
(五)脱氧核糖核酸酶Ⅰ
脱氧核糖核酸酶(DNaseⅠ)来源于牛胰,是一种需要二阶阳离子的内切酶。降解双链DNA产生带3'羟基的寡核苷酸。在Mg2+存在下,在双链DNA上产生缺口,在Mn2+存在下,断裂双链DNA(图1-22)。
1.反应条件
100μL反应体系:
50mmo1/L Tris-HCl,pH7.5
10mmol/L MgCl2(用于产生单链缺口,使双链DNA断裂时用10mmol/L MnCl2代替)
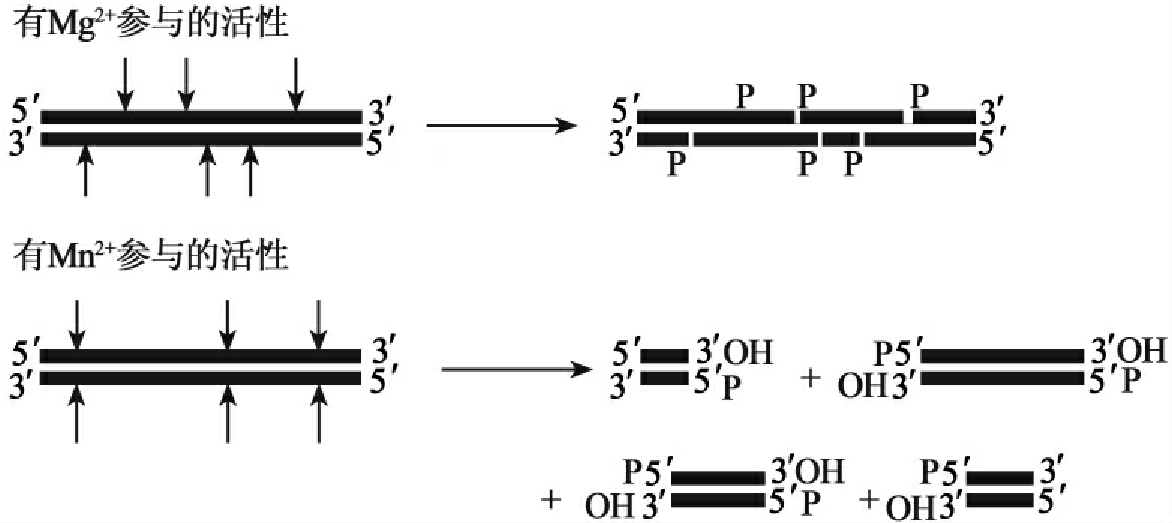
图1-22 脱氧核糖核酸酶Ⅰ(DNaseⅠ)的催化活性
2μg DNA
50μg/mL BSA
1μLDNaseⅠ(其浓度依具体反应而定)
37℃温育1~30min,具体温育时间依消化程度而定。加入5μL 0.5mol/L EDTA终止反应。切口平移反应时,DNaseⅠ的反应与DNA聚合酶Ⅰ的反应同时进行。
2.制备和贮存DNaseⅠ溶液
商品化的DNaseⅠ是一种冻干的粉末(2000~3000U/mg蛋白质),配成溶液后保存期达1年以上。用1mL含20mmol/L Tris-HCl,pH7.5和1mmol/L MgCl2的50%(m/v)甘油溶解1mg DNaseⅠ(不需振荡助溶),-20℃保存。另外,也可用1mL含1mmol/L MgCl2的20mmol/L,pH7.5的Tris-HCl缓冲液溶解1mg DNaseⅠ(不需振荡助溶),以10μL的小份分装于微量离心管中,在干冰上迅速冻结,-80℃保存。避免反复冻融。
3.无RNA酶活性的DNaseⅠ的制备
以0.1mol/L碘乙酸、0.15mol/L乙酸钠溶液(pH5.3)溶解DNaseⅠ,浓度为1mg/mL。55℃加热40min后冷却。加1mol/L CaCl2至5mmol/L终浓度,分成小份,冷冻保存。
4.用途
(1)切口平移。标记DNA探针时,可用DNaseⅠ在双链DNA上产生随机切口。(2)在Mn2+存在时裂解双链DNA.,用于随机克隆。
九、核糖核酸酶
核糖核酸酶(RNase)用于RNA测序、RNA作图及RNA定量等。其中核糖核酸酶A用于水解DNA样品中污染的RNA,核糖核酸酶H和核糖核酸酶TⅠ则用于其他方面。
许多商品化的核糖核酸酶都是序列特异性的核酸内切酶。这种性质使得它们用于RNA测序,如上述3种核酸酶与微球菌核酸酶结合用于RNA测序。
(一)核糖核酸酶A
核糖核酸酶A(RNase A)来源于牛胰,是在C和U残基后特异性水解RNA的内切核糖核酸酶。切割发生在嘧啶核苷酸的3'磷酸基和相邻核苷酸的5'羟基之间。反应终产物为嘧啶3'磷酸及末端带嘧啶3'磷酸的寡核苷酸。RNase A的活性可被来源于人类胎盘的特异性抑制剂RNasin抑制。
1.反应条件
RNase A的反应条件很广,且极难失活。在低盐浓度(0~100mmol/L NaCl)下RNase A切割单链、双链RNA,也能切割RNA∶DNA杂合体中的RNA;但NaCl浓度为0.3mol/L或更高时,RNase A就特异性切割单链RNA。去除反应溶液中的RNase A,通常需要蛋白酶K处理,酚反复抽提和乙醇沉淀。
2.无DNA酶的RNase A的制备
用TE缓冲液溶解RNase A至1mg/mL,煮沸10~30min制备无DNA酶的RNAase A,分成小份存于-20℃。
3.用途
(1)在核糖核酸酶保护实验中,对RNA进行定量和作图,与RNA酶TⅠ联合使用。
(2)降解DNA制备物中的RNA分子。
(3)RNA测序。
(4)将双链cDNA的末端修平,与RNase H联合使用。
(5)从DNA∶RNA杂交体中去除未杂交的RNA区。
(6)确定DNA或RNA中单碱基突变的位置。在此方法中,RNA∶DNA杂交体上的单碱基错配可被RNase A识别并切割。通过凝胶电泳分析切割产物的大小即可确定错配的位置。
(二)核糖核酸酶H
核糖核酸酶H(RNase H)是来自大肠杆菌的内切酶,它特异地水解RNA∶DNA杂交体中RNA的磷酸二酯键,产生末端为3'羟基和5'磷酸的产物,它不降解单链或双链的DNA或RNA。RNase H的水解可通过用较短的寡核苷酸与RNA的杂交而限定于特定位点。
1U酶定义为在37℃20min内水解polyA·poly(dT)生成1nmol的酸溶性核苷酸所需的酶量。
1.反应条件
100μL反应体积:
20mmol/L HEPES-K0H,pH8.0
2μg RNA∶DNA杂合双链
50mmol/L KCl
50μg/mL BSA
1mmol/L DTT
1U RNase H
4mmol/L MgCl2
37℃温育20min,加入1μL 0.5mol/L EDTA。反应体积、DNA的量、酶的量、反应温度、时间及终止反应的方法可视具体应用而变。
2.用途
(1)降解cDNA第一链合成时产生的RNA∶DNA双链体中的mRNA分子,以利于双链cDNA的合成。
(2)通过合成脱氧核糖核苷酸产生局部的RNA∶DNA双链体,诱发特异性的RNA降解。
(三)核糖核酸酶TⅠ
核糖核酸酶TⅠ(RNase TⅠ)是来源于米曲霉的内切酶,它特异地作用于鸟嘌呤核苷酸3'端磷酸并切割与相邻核苷酸相连的磷酸二酯键。反应终产物为鸟嘌呤核苷3'磷酸及末端带鸟嘌呤核苷3'磷酸的寡核苷酸。
1.反应条件
RNaseTⅠ的反应条件很广,且极难失活。在低盐浓度(0~100mmol/L NaCl)下RNaseTⅠ切割单链、双链RNA,也能切割RNA∶DNA杂合体中的RNA;但NaCl浓度为0.3mol/L或更高时,RNaseTⅠ就特异性切割单链RNA。去除反应溶液中的RNaseTⅠ,通常需要蛋白酶K处理,酚反复抽提和乙醇沉淀。
2.用途
(1)在核糖核酸酶保护实验中,对RNA进行定量和作图,与RNaseA联合使用。
(2)RNA测序。
(3)确定鸟嘌呤含量低的DNA模板在体外转录中的转录水平。因为RNase TⅠ优先水解那些非特异的转录产物。
(4)去除DNA∶RNA杂交体中未杂交的RNA区。
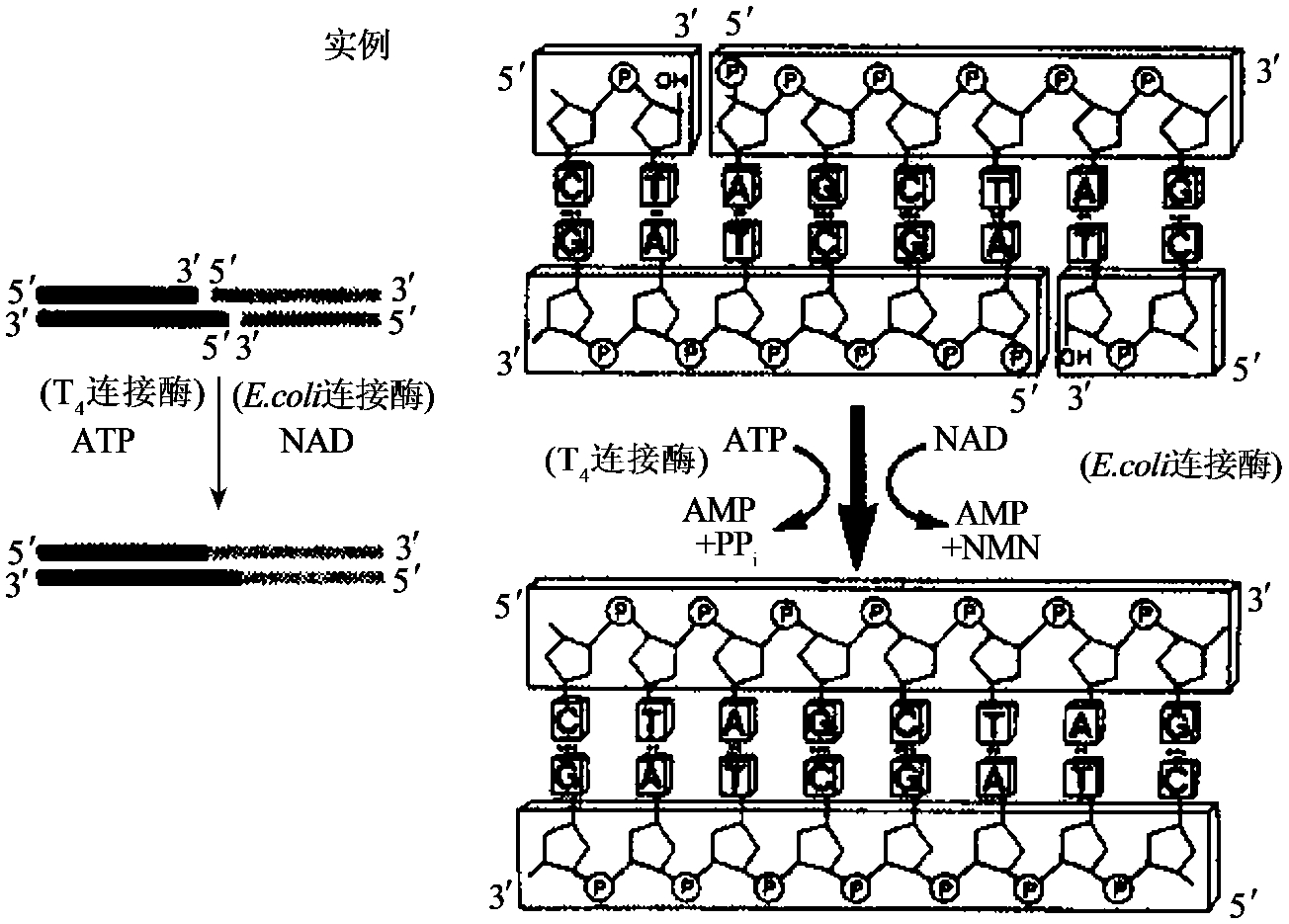
图1-23 DNA连接酶修复单链缺口的活性
十、DNA连接酶
将两段DNA拼接起来的酶称DNA连接酶。它催化双链DNA中相邻碱基的5'磷酸和3'羟基间形成磷酸二酯键。此活性可用于修复双链DNA中的单链缺口(图1-23),连接带平端(图1-24)或带同源黏端(图1-25)的双链DNA限制酶切片段,用于核酸研究的有两种DNA连接酶,即T4DNA连接酶和大肠杆菌DNA连接酶。这两种酶的性质有两点不同:①所用能源不同,T4DNA连接酶以ATP为能源,大肠杆菌DNA连接酶则以NAD为能源;②正常反应条件下,只有T4DNA连接酶能连接平端,而大肠杆菌DNA连接酶则不起作用。因此分子克隆中最常用的为T4DNA连接酶。
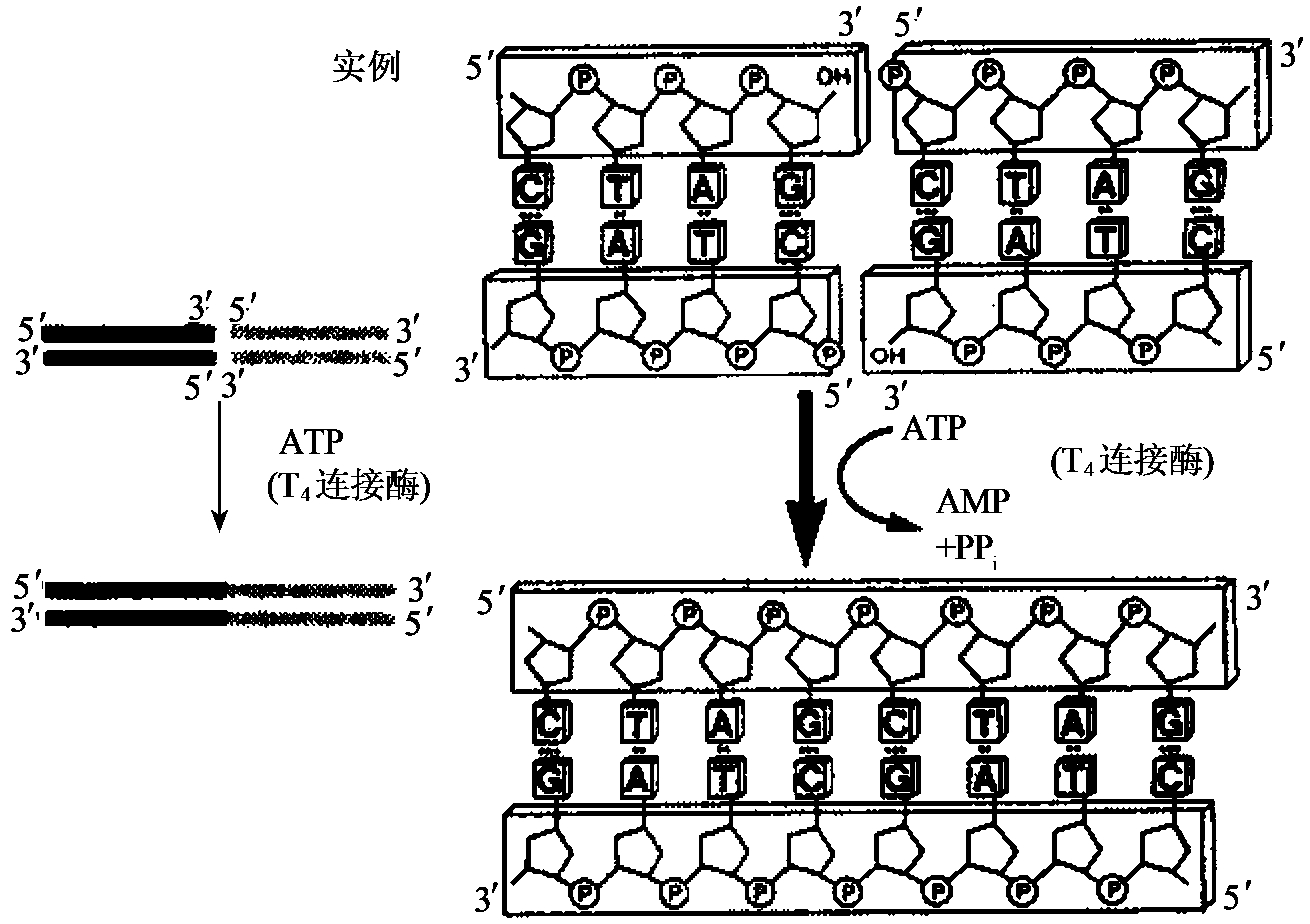
图1-24 DNA连接酶连接平端DNA的活性
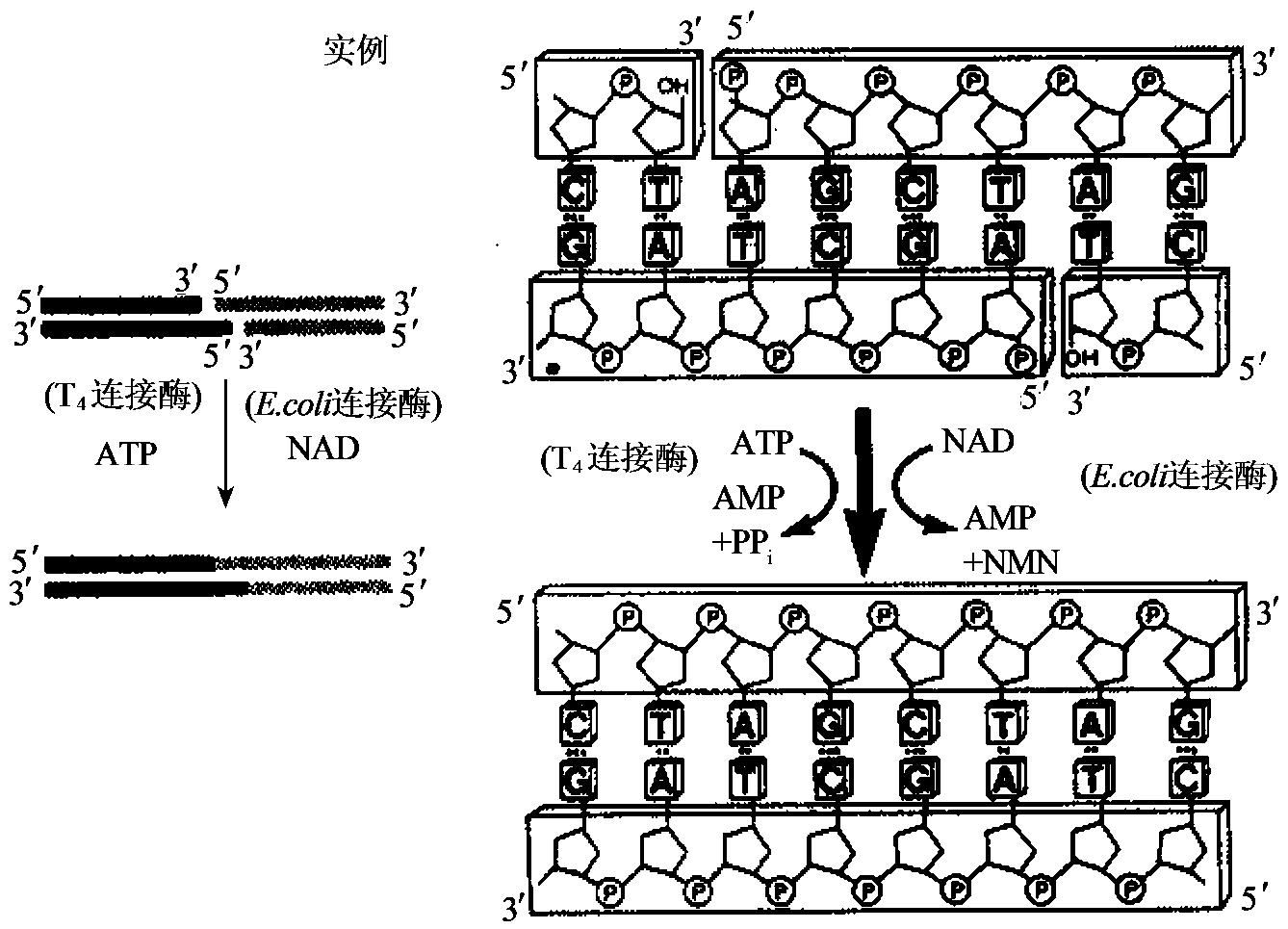
图1-25 DNA连接酶连接黏端DNA的活性
(一)T4DNA连接酶
此酶为T4噬菌体基因30的产物,由一条多肽组成,分子质量为68kU。目前已经通过基因工程方式制备并销售。该酶能催化两条DNA链的3'羟基和5'磷酸之间形成磷酸二酯键而把两个DNA分子连接在一起。它不但能把两个具有黏性末端的DNA分子连接在一起或把一个DNA分子的两个黏性末端连接起来使之成为环状,而且也能把具有平端的DNA分子连接起来。连接两个DNA分子的先决条件是它们的末端必须靠拢在一起,所以连接黏性末端的分子比连接平端分子的效率要高得多。目前对平端连接的过程尚不清楚,有人认为平端的连接至少需要两个连接酶分子。其中一个分子握住两个平端使它们维持靠拢状态,另一分子催化磷酸二酯键形成。所以在进行平端连接时需要DNA的浓度更高和更多的连接酶。曾报道用8%~16%的PEG 8000可大大提高平端连接和黏端连接的效率。另外加入T4RNA连接酶对平端的DNA连接也有促进作用。PEG在这里可起占据体积的作用,从而有效地提高了平端DNA及酶的浓度,可增强水溶液中大分子间的有效作用。
TDNA连接酶催化反应必须存在Mg2+和ATP,最适pH值为7.5~8.0。就酶活力本身4而言,温度在37℃时活力最高,5℃以下活力显著降低。由于必须考虑DNA的稳定性和黏性末端的退火温度,黏性末端的连接通常在12~15℃进行,平端的连接通常在室温(<30℃)进行,所需酶量是黏端连接的10~100倍。平端连接反应比黏性末端连接慢得多,但单价阳离子(低于150mmol/L NaCl)及低浓度的PEG可大大提高平端连接的速率。在NaCl浓度高于150m mol/L时受抑制。
1.反应条件
50μL反应体系:
40mmol/LT ris-HCl,pH7.5
10mmol/L MgCl2
10mmol/L DTT
1μg DNA
0.5mmol/L ATP
50μg/mL BSA
1“Weiss”U T4DNA连接酶
12~30℃温育1~16h,加入2μL 05mol/L EDTA或75℃加热10min终止反应。反应体积、DNA浓度、反应温度和反应时间依具体应用而变。酶的单位为Weiss单位,1个Weiss单位相当于60个黏性末端单位。
2.用途
(1)连接带匹配黏端的DNA分子。
(2)使平端的双链DNA分子互相连接或与合成接头相连接。
(二)大肠杆菌DNA连接酶
大肠杆菌DNA连接酶是lig基因编码的产物,现已通过基因工程方式克隆并表达,该酶可修复双链DNA中的单链切口并可催化互补黏性末端的连接。PEG 8000可增加黏端的连接效率,亦可促使该酶有效地连接平端。在正常反应条件下,它不能连接平端,但是,在聚乙二醇(PEG)或聚蔗糖(Ficol1)存在下,该酶能催化平端连接。聚乙二醇和聚蔗糖在这里起占据体积的作用,从而有效地提高了平端DNA和酶的浓度。由于合成cDNA第二链时出现的RNA和DNA不会被大肠杆菌DNA连接酶连接,因此用置换合成法做cDNA克隆时可使用大肠杆菌DNA连接酶。但在其他分子克隆操作时该酶并不常用,这是因为T4DNA连接酶在没有体积占据时也能有效地连接平端。该酶活性以Modrich-Lehman单位表示,1个Modrich-Lehman单位相当于6个Weiss单位。
1.反应条件
50μL反应体系:
40mmol/L Tris-HCl,pH8.0
10mmol/L MgCl2
5mmol/L DTT
1μg DNA
0.1mmol/L NAD
50μg/mL BSA
10“Modrich-Lehman”U大肠杆菌DNA连接酶
10~25℃温育2~16h,加入2μL 0.5mol/L EDTA或75℃加热10min终止反应。反应体积、DNA浓度、反应温度和时间依具体应用而变。
2.用途
大肠杆菌DNA连接酶在非平端连接时可代替T4DNA连接酶,它产生的背景低,准确性高。
十一、RNA连接酶
T4RNA连接酶是T4噬菌体基因63的编码产物。在ATP存在下,它催化单链DNA或RNA的5'端磷酸与单链DNA或RNA的3'端羟基共价连接(图1-26)。
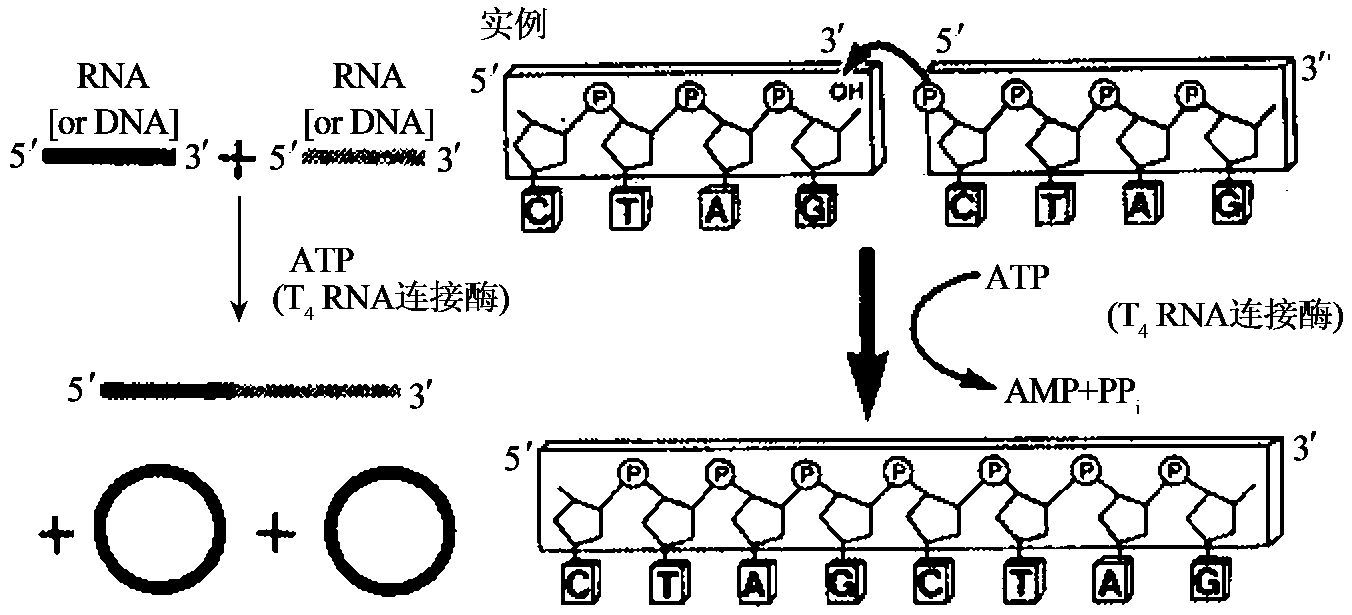
图1-26 RNA连接酶活性
1.反应条件
50μL反应体系:
50mmol/LHEPES,pH8.3
10mmol/L MgCl2
5mmol/L DTT
2μg单链DNA或RNA
2mmol/L ATP
50μg/mL BSA
1UT4RNA连接酶
17℃温育10h,加入2μL 0.5mol/L EDTA终止反应。
2.用途
(1)RNA 3'末端的放射性标记。在标记反应中,RNA为受体,5'32P标记的3',5'-二磷酸核苷(如5'-32P Gp)为供体,ATP为能源。反应终产物为含有标32P标记的3'末端核苷酸的RNA分子。
(2)寡聚DNA和RNA分子环化。
(3)在寡核苷酸合成中,连接寡聚物产生在特异性位点上带有内部标记的寡聚体。
(4)增加T4DNA连接酶的平端连接活性。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。











