



第三节 功能基因组学
随着基因组的研究从结构基因组向功能基因组转换,一个重要的挑战是如何从已完成测序的基因组中获取大量有用的信息来解释基因的功能、特定时空下细胞或组织中基因的表达情况以及相互调控的过程,以研究及了解生物体的特性,其中,基因表达水平的分析起着重要的作用。
目前,用于基因表达水平研究的主要方法有:DNA微阵列、EST序列鉴定和基因差异显示法等。但他们都有一定的局限性,如DNA微阵列只能对已知基因的表达进行检测,不能发现未知基因;EST序列鉴定虽然能发现新基因,但是它的检测量有限、工作效率低;基因差异显示法只能反映有限的基因表达频谱,无法描述基因表达的全貌,且都针对高丰度或表达差异大的基因,但细胞中表达的绝大多数基因属于低丰度表达,且相当一部分具有重要功能的未知基因。
一、SAGE技术
SAGE技术是在1995年由Velculescu等首先提出来的,是一种快速分析基因表达信息的技术。它是以获得的基因组数据库为依据,通过快速和详细地分析成千上万个EST来寻找出表达丰度不同的SAGE标签序列(tag)以了解生物体中基因的表达类别及其丰度。该技术不仅对低丰度表达的基因有较好的检测效果,而且还能发现新的基因。
1.SAGE的原理
(1)在一个转录体系中,每种转录本都可以用一段特异的固定长度的短标签(SAGE tag)来代表,该短标签包含了足够的特异信息来确定该转录本。以一个9bp的标签序列为例,理论上可以区分49(262144)种转录本,而人类基因组估计仅编码80000种转录本,所以9bp足以鉴别任何一种特定的人组织或细胞中的全部转录本。
(2)将SAGE标签分离出来,连接形成串联体,克隆到载体内进行扩增、测序分析,得到代表转录本信息的标签序列,经过数据处理后获得代表标签序列的基因信息及其表达丰度。
2.SAGE的研究方法 SAGE的主要操作分为3个阶段(图12-3)。
第一阶段:SAGE文库的构建。
(1)提取细胞总RNA或mRNA,以生物素标记的寡核苷酸(Oligo-dT)为引物,反转录合成ds-cDNA,用锚定酶(NlaⅢ)酶切生物素化的ds-cDNA,该酶能够识别CATG位点并在其3′侧进行酶切。锚定酶要求至少在每一种转录本上有一个酶切位点,一般用4碱基限制性内切酶能满足这个要求。酶切产物用链霉亲和素包被的磁珠收集3′端ds-cDNA酶切片段。
(2)分离到的cDNA的5′端补平,并分成2份,分别连接接头A或接头B,每种接头都含有标签酶识别位点(常用BsmFI)、黏性末端(与NlaⅢ酶切后留下的凸端GTAC互补,即锚定酶酶切位点)以及一个PCR引物的序列,长度为40bp核苷酸,连有接头的cDNA用BsmFI酶切,该酶在其识别位点3′端下游的14~15bp处进行切割,从而使每条cDNA释放出一个带有接头的短cDNA片段(9~10bp)。
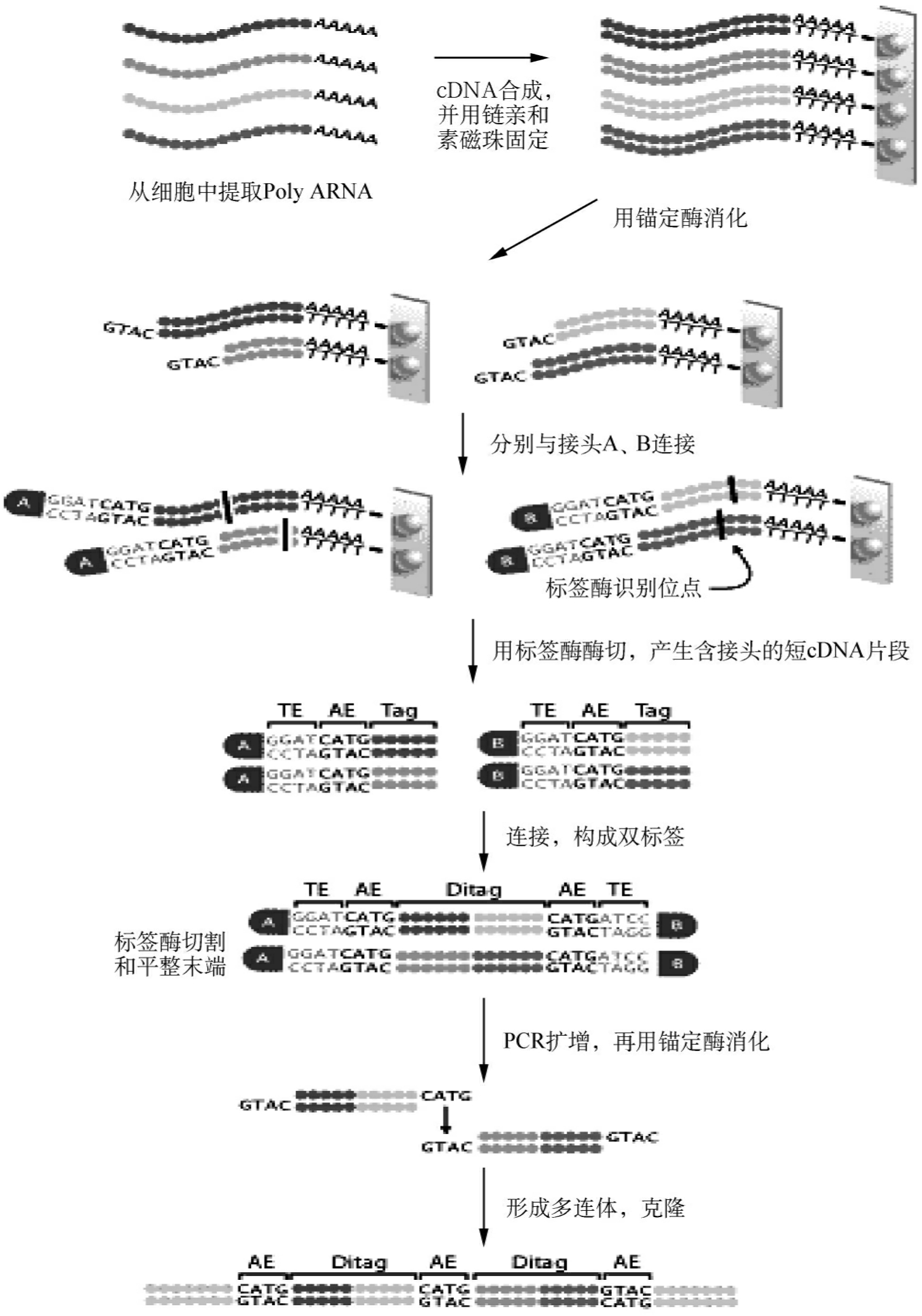
图12-3 SAGE的主要操作过程
(3)使用Klenow反应补平酶切后的5′黏性末端。将含有接头A和接头B的cDNA短片段混合,用DNA连接酶进行连接,形成100bp的双标签片段,使用引物A和引物B对双标签片段进行PCR反应,然后用锚定酶NlaⅢ酶切扩增产物,释放出连接接头,分离纯化双标签片段,用DNA连接酶连接双标签片段形成串联体,串联体的长度为300~1000 bp,收集串联体克隆到高拷贝的克隆载体中。
第二阶段:SAGE文库的测序。利用克隆载体的通用引物,对插入的串联体进行单向测序,收集资料。测序时原始数据的质量非常重要,同时要求测得的片段尽可能长。因为错误的单碱基序列会导致原有标签的有用信息被丢失,从而产生一个并不存在的标签。
第三阶段:数据的分析。数据分析通常使用SAGEnet提供的软件包或者通过在线的NCBI来完成。首先从串联体的原始资料中定位NlaⅢ的酶切位点(即CATG),提取CATG位点之间的20~26bp的双标签序列,然后去除重复出现的双标签序列,包括在反向互补链上的重复双标签序列,去除与接头序列相对应的标签,同时去除含有不确定碱基的标签。通过与基因数据库资料的分析比较,将这些SAGE标签确定为特点的转录子,并计算每个标签的出现次数来表示其在转录体中的丰度。
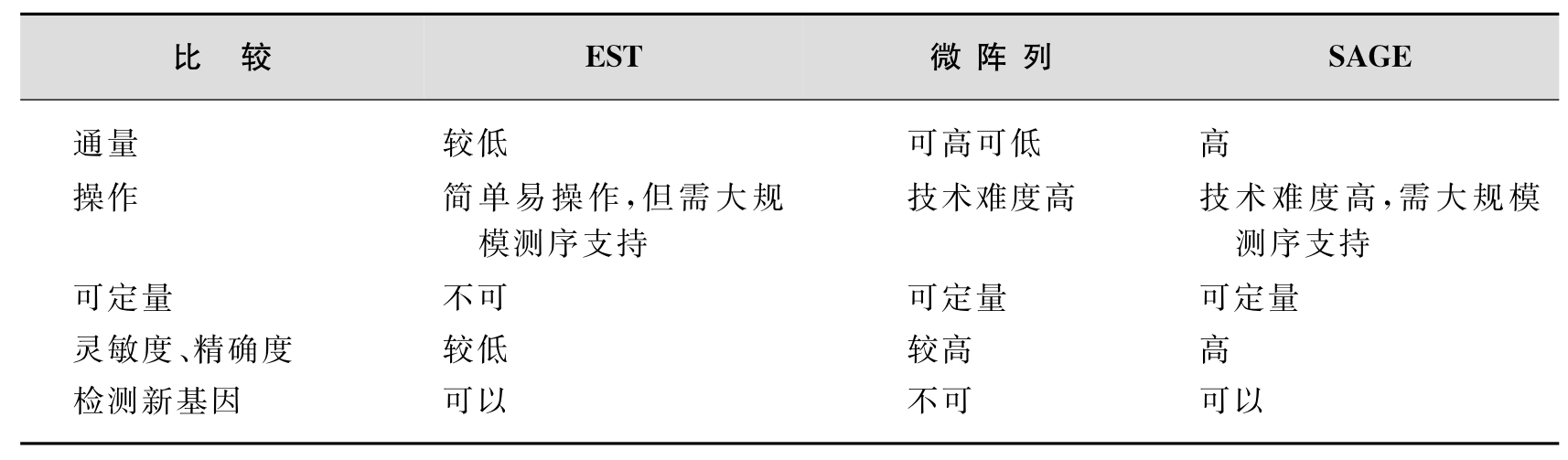
3.SAGE技术方法的改进 虽然SAGE有许多优点,如分析低丰度转录本的表达情况、可以发现新基因等,但是它也有着需要的样本量多、技术流程复杂、工作量大以及标签的确认困难等缺点。针对这些情况出现了一些改进的SAGE技术(表12-2)。
表12-2 SAGE与相关技术的比较
(1)microSAGE:经典的SAGE需要的样本量大,5~50μg总RNA或50~500ng mRNA。不适于微量组织的研究。1999年,Datson提出了微量SAGE(microSAGE)的方法,只需要常规SAGE1/5000~1/500的样本量,有利于研究一些来源有限的组织的基因表达分析。同时,microSAGE从RNA的提取到获得标签的整个操作过程均在同一试管内进行,提高了工作效率,减少了纯化步骤。
(2)Long SAGE:SAGE技术理论上可以鉴别所有的基因,但在实际应用中还是存在一些问题。人们在分析SAGE标签时,出现了许多多重匹配标签或者未知标签,因而不能确定这些标签在组织中的基因表达情况。这是由于SAGE技术得到的标签长度太短,这些短标签所携带的信息量在复杂的数据库中不利于对相关基因的确认。因此,在2000年由Ryo等建立了Long SAGE技术,他们使用限制性内切酶RsaⅠ(识别位点为GATC)作为锚定酶,通过构建一个非回文结构的黏性连接子(A-T连接),用BsmFI作为标签酶,酶切后可得到一个14bp长度的标签序列(图12-4),14bp的标签减少了多重匹配和未知标签的出现,特异性明显提高。
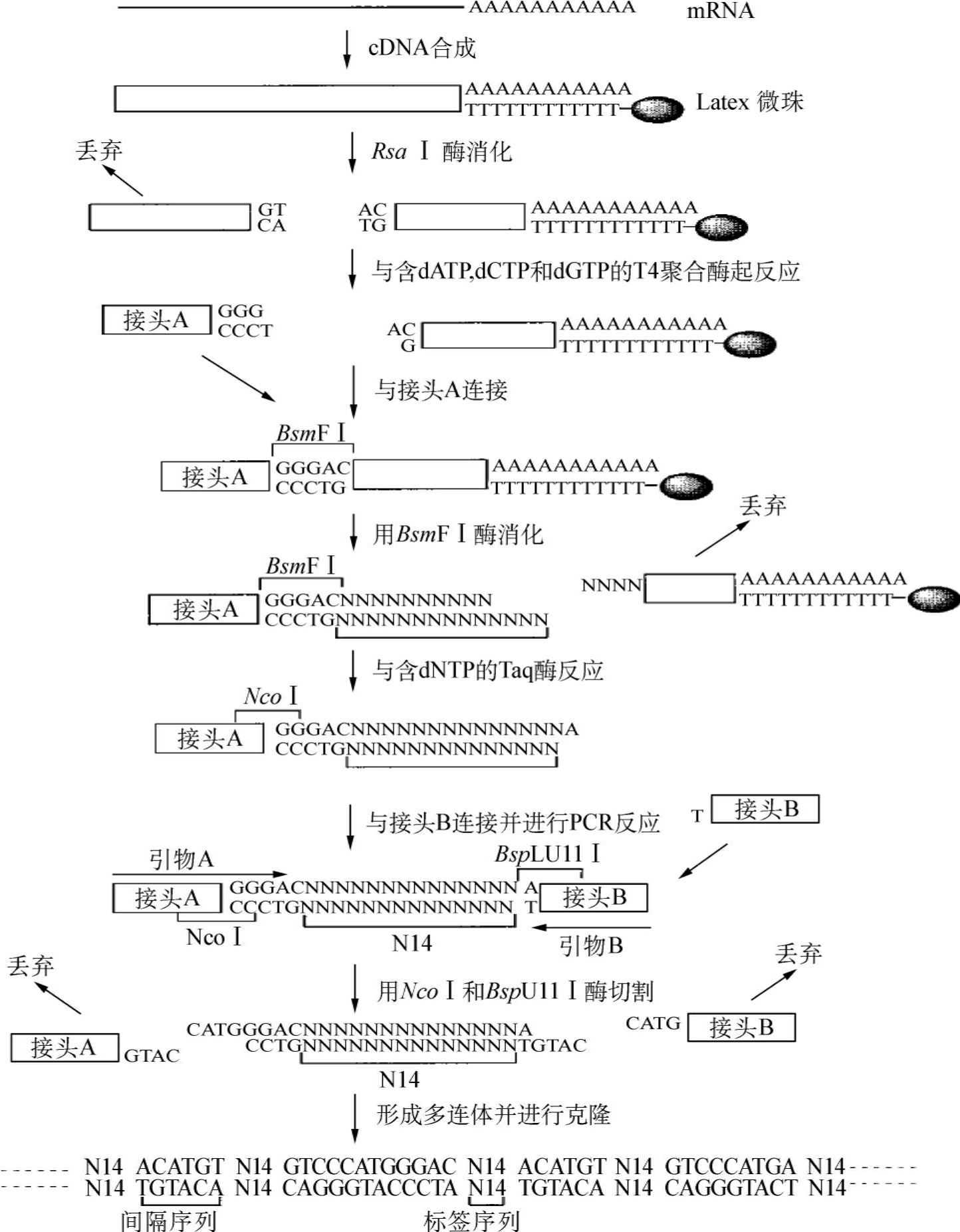
图12-4 Long SAGE技术
二、RNAi文库技术
RNA干扰是一种古老的天然抗病毒机制,在动物界和植物界普遍存在,它是双链RNA分子在mRNA水平上诱发的序列特异性的转录后基因表达沉默。从1998年以来,它的理论机制和技术应用取得重大进展。RNAi的可能机制如下:长片段dsRNA在细胞胞质内被RNaseⅢ型样酶Dicer切成长度为21~23nt的小片段干扰RNA(small interfering RNA,siRNA),这些双链siRNA调入到Argonaute2(Ago2),构成RISC(RNA-induced complex)复合物的催化单位。siRNA通过与同源mRNA的特异配对,引导RISC特异地降解同源mRNA,导致基因表达的抑制。因此,小片段的siRNA也可以诱导高效的基因沉默。
随着人类基因组的完整序列的获得,破译基因组内所包含信息的工作才刚刚开始。目前,大多数基因在生物体内的功能还不清楚,而传统研究基因功能的方法(如基因敲除技术等)由于技术复杂,操作周期长,费用高等缺点,使得基因功能的研究进展缓慢。而RNAi技术是一种高效、特异、易操作的基因阻断技术,为大规模基因功能的研究提供了有力的武器。
RNAi文库(RNAi library)是人工构建的能通过诱导RNAi抑制众多不同基因表达的混合文库,可以用于建立功能缺陷(loss of function)的生物或细胞库,进行表型的筛选。该技术在线虫的应用中最为成功。在线虫等模式生物的研究中,应用RNAi文库建立随机基因抑制的线虫,再进行表型的筛选,已在其胚胎发育等领域取得了重要的发现。
通过从基因组水平设计构建RNAi文库进行系统性、大规模的基因功能缺陷(loss of function,LOS)筛选,是研究功能基因组学的重要武器,为发现新的药物靶基因、肿瘤的形成和发展等研究提供了重要的方法。应用RNAi技术在基因组范围内筛选功能基因时,必须仔细地评估构建的RNAi文库的基因覆盖率,以便提高基因筛选的效率和数据的可重复性。按照文库构建的方法,大致可分为以下几种:化学合成的文库、质粒(或病毒)的文库、siRNA表达盒文库和随机RNAi文库。
1.化学合成的文库 一开始构建RNAi文库时,只针对线虫、果蝇等无脊椎动物,通过体外转录生成1~2kb的dsRNA片段来构建,在低等生物中,这些长的dsRNA具有很高的特异性,并能有效地诱导基因沉默。但是在哺乳动物等高等动物中,由于存在着抗病毒机制,长片段dsRNA会引起细胞的毒性反应而失活,直到人们发现用短片段(21nt)的siRNA能避免抗病毒机制,有效地引起基因功能缺陷,才使得人们通过化学合成的方法,将RNAi技术在高等生物中得到应用。
Aza-Blanc等建立了包含针对510个人类基因(多数为激酶基因)的siRNA寡核苷酸文库,他们通过转染HeLa细胞,用以筛选TRAIL诱导凋亡信号转导途径的调节基因,结果除检测到已知的该途径的调节基因外,还发现bobi、mirsa、myc、wnt、jnk以及bmk1/erk5与TRAIL凋亡途径相关。但是因为化学合成RNA成本太高,又无法扩增,RNA易被降解,只能获得短暂的抑制效果,使得这类文库的应用范围受到限制。
2.质粒(或病毒)的文库 质粒(或病毒)的文库是由大量带有特异性目的基因编码序列的质粒或病毒构成,将其导入细胞后,可表达针对特异基因的shRNA(small hairpin RNA)或siRNA(small interfering RNA),从而达到基因沉默的效果。由于构建的是已知基因的文库,其工作量较大,刚开始时,只是针对几百个基因的小容量的文库,以后逐渐扩大到数千个基因的siRNA文库。如Bern等利用反转录病毒载体pROTRO SUPER构建了针对7914个人类基因的RNAi文库;Paddison等利用另一种反转录病毒载体pSHAG构建了针对9610个人类基因和5563个小鼠基因的RNAi文库。这类文库的优点是可以获得较高的转染效率(尤其是病毒载体),并可实现稳定表达,文库可以扩增,再生性较好,比较适合长期筛选的实验;该文库还可以不断扩充新的成员,文库的成员可以分开也可混合使用,因此既可进行混合库形式的筛选,也可进行独立单克隆矩阵形式的筛选。
3.siRNA表达盒文库 在低等生物中,用化学合成或mRNA裂解形成的siRNA能很好地行使基因沉默是因为它存在内源性的放大机制,但在哺乳动物中,由于不存在内源性的放大机制,化学合成的RNA分子,只能获得短暂的目的基因活性下调,对于大多数的高通量实验筛选(highthrough screening,HTS)短暂的下调是足够的,但对于需要基因长期沉默来观察生物学行为的实验,短暂的基因下调是不够的。因此,人们开始设计适用于高等生物细胞内表达siRNA的分子。
siRNA表达盒(siRNA expression cassette,SEC)是一段PCR产物,它依次包含RNA聚合酶Ⅲ依赖的启动子、靶基因的反义链、loop环、正义链和转录终止序列,将其转染细胞可直接转录出完整的双链siRNA,发挥基因表达沉默的效应。
Zheng等构建了一个双启动子载体pDual。该载体包含两个方向相对的聚合酶Ⅲ启动子,分别为小鼠的U6启动子和人的H1启动子,中间插入能转录siRNA的DNA序列。正义和反义RNA从同一DNA序列转录,减少了合成DNA序列的长度。他们用PCR扩增的含双启动子的siRNA表达盒,证实了可以在哺乳动物细胞中有效地抑制基因的表达。应用这一技术,他们构建了针对8000个基因的siRNA表达盒,在大规模的功能基因筛选中发现了一些已知和未知的在NF-κB信号转导通路中起重要作用的基因。Root等用修饰的慢病毒(PLKO.1lentiviral vector)作为载体构建了一个覆盖了14500个人的基因和12 700个小鼠基因的shRNA文库,该文库的特点是筛选的重复性高、稳定性好,在分裂细胞和非分裂细胞中都能特异地抑制目的基因的表达,使用抗性基因能够有效地筛选阳性克隆。用RT-PCR的方法检测针对59个基因的283个克隆shRNA的基因沉默效率,平均为47%;其中38%的shRNA的基因沉默效率超过70%,使用8种shRNA的3个不同的剂量,未见到非特异性细胞毒性反应。
由于这种方法不需要经过载体克隆筛选和测序等繁琐的步骤,只需将PCR产物转染到细胞中就可对不同的siRNA进行快速筛选。不需要体外转录,避免了复杂的RNA操作,同时又比化学合成法成本低。因此它具有高效、快速、相当廉价和易操作等优点,已经被用于RNAi文库的构建。
4.随机RNAi文库 已知基因RNAi文库构建时必须知道靶基因的序列,由于不是随机的,文库的覆盖率低,为了获得较好的干扰效果,常需要构建不同的siRNA,所需的费用高,而且筛选的工作量大。随机RNAi文库(random RNAi library)是人工构建的可能针对任何基因的RNAi文库,因此文库的容量大,克服了已知基因RNAi文库细胞基因覆盖面窄的缺陷。
随机RNAi文库也称为酶切消化产生的RNAi文库,它是利用特定的酶(限制性内切酶MmeⅠ)切割基因组DNA或cDNA,得到一系列的随机DNA片段,克隆到相应的载体上,构建而成的文库。限制性内切酶MmeⅠ的特点是在DNA识别序列(TCCGAC)下游20~21bp处切开DNA片段。
Luo等应用MmeⅠ的独特特性,设计了将长cDNA片段酶切为20~21nt小DNA片段,克隆于siRNA表达载体构成随机RNAi文库的方案,称为SPEED(small interfering RNA production by enzymatic engineering of DNA)。首先将cDNA用DNaseⅠ或者限制性内切酶切割成许多含有黏性末端的cDNA片段,与含MmeⅠ酶切位点发夹结构的DNA接头进行连接,再用MmeⅠ消化,获得20~21bp与接头连接的cDNA小片段。将形成的ds DNA打开成ss DNA,合成互补链,形成方向相对的两个小片段cDNA的拷贝,中间为不配对的形成发夹结构的DNA序列。再克隆到MSCV-pgk-EGFP反转录病毒载体中,构成随机RNAi文库。利用小鼠胚胎cDNA文库,他们建立了含有3×106克隆的RNAi文库,随机挑选27个克隆序列插入的片段,证实了它们是小鼠的已知基因,这些克隆在小鼠NIH3T3细胞系中能稳定表达,他们用RT-PCR分析了548个克隆,发现在细胞中明显抑制了相应基因的mRNA表达,证明该方法的可行性。Shirane等应用同Luo等基本相同的思路独立地设计了构建随机RNAi文库的方法,称为EPRIL(enzymatic production of RNAi libraries form cDNA)。为了快速筛选阳性克隆,他们在插入片段前加入一个筛选基因(由胸腺嘧啶激酶和呤N-乙酰转移酶组成的融合蛋白),通过更昔洛韦(ganciclovir)获得阳性克隆。应用小鼠髓样祖细胞(FL5.12细胞)的cDNA文库,建立了随机RNAi文库。对随机选择的240个不同克隆的测序,发现215个克隆含有正确的能表达siRNA的DNA片段。BLAST分析显示,180个克隆的插入片段中有165个与已知基因或EST的顺序相吻合,其中146个属于UniGene。这一结果说明,EPRIL可以用于复杂基因的随机RNAi文库的构建。Sen等独立地设计了相似的建库方案,称为REGS(restriction enzyme-generated siRNA)。为了使每个基因获得多个siRNA,用多种限制性内切酶(HinpⅠ、BsaHⅠ、AciⅠ、Hpy、ChiⅤ和TaqαⅠ6种RE)消化cDNA文库构建RNAi文库。他们用该文库检测了对内源基因的作用,可以有效地抑制胚胎干细胞中Oct-3/4的表达,使得ES细胞朝着滋养层方向分化,因为Oct-3/4基因的表达对于胚胎干细胞的自我更新起着重要作用(图12-5)。
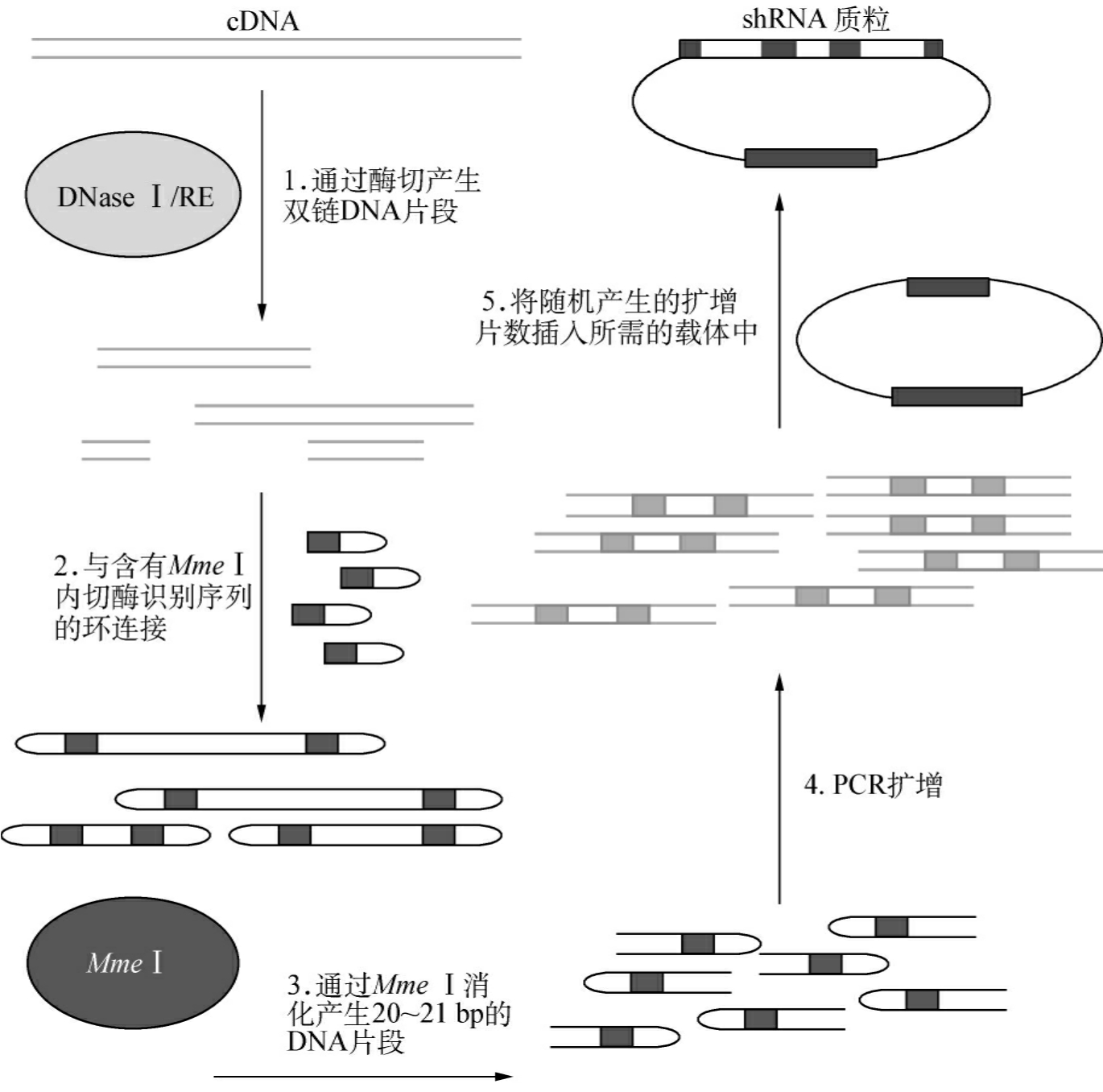
图12-5 随机RNAi文库
引自Julie Clark and Sheng Ding.Generation of RNAi 2006,45716:1-7
miRNA是一类内源性的dsRNA,通过RNAi途径产生生物效应。第一个miRNA(lin-4)是在线虫中发现的,随着时间的推移,人们逐渐认识了这类非编码调控RNA的生物合成、加工和调节功能。miRNA是由RNA聚合酶Ⅱ转录,生成一条长的含有PolyA的初级转录产物(pri-miRNA),在细胞核中被RNaseⅢDrosha切割成为前体miRNA(pre-miRNA)。在最初的剪切后,pre-miRNA在转运蛋白exportin-5的作用下由核内转移到胞质中,然后由另一种RNaseⅢ(Dicer)进一步切割产生成熟的miRNA,这些成熟的miRNA与其他蛋白质一起组成RISC复合体,从而引起靶mRNA的降解或者翻译抑制。Chang等根据RNAi的生物特性和miRNA的代谢途径构建以miRNA为基础的人和小鼠的第2代基因组shRNA文库,该文库具有更高的基因沉默效应,它比第一代基因组shRNA文库的基因沉默效率高12倍,并且可应用于组织特异性的表达系统中。
到目前为止,针对哺乳动物全基因的RNAi文库的构建还存在一些问题需要克服,但该技术已经在哺乳动物大规模基因功能缺失的筛选和研究中显示出巨大的潜力,为促进功能基因组学的研究提供了一种有效的工具。
(吴春根 张 彦)
参考文献
1.坎贝尔·A马尔科姆主编.孙之荣主译.探索基因组学、蛋白质组学和生物信息学.北京:科学出版社,2005
2.丹尼斯C,加拉格尔R主编.林侠等主译.人类基因组我们的DNA.北京:科学出版社,2003
3.杨金水主编.基因组学.北京:高等教育出版社,2002
4.本杰明,卢因主编.余龙等主译.基因Ⅷ.北京:科学出版社,2005
5.颜子颖等主译.精编分子生物学实验指南.北京:科学出版社,1998
6.张丽妹等.RNA干扰在哺乳动物细胞高通量基因功能研究中的应用.医学分子生物学杂志,2005,2(5):355~358
7.邓永键等.基因表达系统分析及其应用前景.免疫学杂志,2003,19(4):315~321
8.张莹等.RNA干扰文库在功能基因组学研究中的发展及应用.中国生物工程杂志,2006,26(7):84~89
9.Aza-Blanc P,et al.Identification of modulators of trail-induced apoptosis via RNAi-based phenotypic screening.Molecular Cell,2003,12:627~637
10.Berns K,et al.A large-scale RNAi screen in human cells identifies new components of p53pathway.Nature,2004,428:431~437
11.Mostafa Ronaghi,Pyrosequencing sheds light on DNA sequencing.Genome Res.2001,11:3~11
12.Nader Poumand,et al.Multiplex Pyrosequencing.Nucleid Acids Res.2002,30(7):31~35
13.Rene H.Medema,Optimizing RNA interference for application in mammalian cells.Biochem J,2004,380:593~603
14.Julie Clark and Sheng Ding.Generation of RNAi Libraries for High-Throughput Screens.J Biomede Biotechnol.2006,(457)16:1~7
15.Akihide Ryo,et al.A Modified Serial analysis of gene expression that generates Longer Sequence Tags by nonpalindromic cohesive linker ligation.Anal Biochem.2000,277:160~162
16.Chan Ho Song,et al.Painless gene expression profiling:SAGE(Serial analysis of gene expression).The Science Creative Quarterly August,2004
17.Luo B,et al.Small interfering RNA production by enzymatic engineering of DNA(SPEED).PNAS,2004,101(15):5494~5499
18.Paddison P J,et al.A resource for large-scale RNA-interference-based screen in mammals.Nature,428:427~431
19.Robert E,et al.Pyro Q-CpGTM:quatitative analysis of methylation in multiple CpG sites by Pyrosequencing.Nat Methods,2005,oct i~ii
20.Root DE,et al.A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high content screen.Cell,2006,126(6):1283~1298
21.Silva JM,et al.Second-generation shRNA libraries covering the mouse and human genome.Nat Genet,2005,37(11):1281~1288
22.Shirane D,et al.Enzymatic production of RNAi libraries from cDNAs.Nat Genet,2004,36(2):190~196
23.Sen G,et al.Restriction enzyme-generated siRNA(REGS)vectors and libraries.Nat Genet,2004,36(2):183~189
24.Zhen L,et al.An approach to genomewide screens of expressed small interfering RNAs in mammlian cells.PNAS,2004,101:135~140
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。









