



7.2.3 人Mn-SOD启动子的突变导致人Mn-SOD在癌细胞中表达减少
Mn-SOD在抑制癌症的发展中发挥重要作用。在许多转化细胞和肿瘤组织中,Mn-SOD的活性都有所减弱。以往已经证明了癌细胞中Mn-SOD活性的减弱不是因为Mn-SOD蛋白的初级结构的缺失,而是由于基因表达的缺陷。为了阐明癌细胞中Mn-SOD表达的减少,研究了正常人体细胞系和各种人类肿瘤细胞系中Mn-SOD基因的调节区域的核苷酸序列。从正常人体基因组DNA库中分离出Mn-SOD基因5′端序列,它是一个3.4kb的基因片段,该基因片段被用来测定Mn-SOD启动子的DNA序列。用PCR引物扩增癌细胞中Mn-SOD基因的5′端3.4kb大小的基因片段。序列分析鉴定了在五种人体肿瘤细胞系的启动子的邻近区域中出现三种杂合突变体。这些突变体,位于人的Mn-SOD启动子的GC富含区,能够改变AP-2的结合方式,并且采用荧光素酶报告基因分析系统发现这些突变体还能导致转录活性降低。这些结果表明在一些肿瘤细胞中Mn-SOD表达的减少,至少部分地,是由于启动区的基因序列的缺失。
积累的数据表明Mn-SOD构成一个主要的细胞防御机制,抵御能够产生氧化压力的试剂的毒副作用。已经证明Mn-SOD抑制小鼠心肌病和新生儿致死,而Cu,Zn-SOD和EC-SOD同功酶基因独立的分解导致正常小鼠在无压力条件下能够存活。此外,氧诱导的肺损伤、急性阿霉素导致的心肌损伤以及局部缺血导致的脑损伤都能够阻止转基因小鼠在线粒体中表达人Mn-SOD。
利用基因转染进行的许多研究已表明Mn-SOD转染到肿瘤细胞能够逆转恶性表型的肿瘤细胞,表明Mn-SOD的功能能够抑制细胞变成肿瘤细胞。人Mn-SOD基因导入小鼠成纤维细胞可防止辐射诱发的恶性转化。在小鼠C3H10T1/2细胞中人类Mn-SOD基因的表达能够增强采用5-阿糖胞苷治疗细胞分化的效果。人黑色素瘤细胞中的呈现恶性表型的细胞能够被6号染色体抑制,而在6号染色体上存在Mn-SOD基因或者转染的人Mn-SOD cDNA。Mn-SOD的过度表达能够抑制人类乳腺癌细胞,人类神经胶质瘤细胞和小鼠表皮细胞的恶性表型。在同源小鼠中Mn-SOD-Fsa-Ⅱ与昆虫媒介者转染的控制细胞相比,前者所需的用来产生肿瘤的细胞数量明显有所增加,而且转移的频率也减少。此外,当活体内Mn-SOD-Fsa-Ⅱ在含氧量低的条件下被移植和辐射时,控制半数组织培养出现的细胞病变量的辐照剂量大大降低。上述研究得出的证据支持由Oberley提出的假说,即Mn-SOD在预防癌症的发展中起着重要的作用。
已经表明人类癌症细胞的许多类型相比正常细胞能够降低Mn-SOD的活性。在人类癌细胞中,Mn-SOD活性的下降不是由于Mn-SOD蛋白的初级结构的缺失,也不是因为Mn-SOD基因剂量的改变或是肿瘤细胞中Mn-SOD mRNA稳定性的降低,而是由于基因表达的缺失。整个人Mn-SOD基因已被克隆和测序,其中包括一个0.78kb的5′端区域,这是从正常的人肺成纤维细胞的基因组文库获得的。该基因的特点是缺乏TATA或CAAT盒,而出现一个包含多重SP-1结合位点的GC富含区。为阐明在肿瘤细胞中人类Mn-SOD表达减少的原因,进一步测序了人Mn-SOD基因的5′端区域并与来自几种肿瘤细胞系的人类Mn-SOD基因的5′端区域进行比较。结果证明人类Mn-SOD基因启动区的三种杂合突变体在14种肿瘤细胞系中的五种中被检测出来。三种突变体对人Mn-SOD启动子的转录活性的影响也被确定。
7.2.3.1 人Mn-SOD基因的5′端区域的克隆和鉴定
从人的肺成纤维细胞系中分离出人类Mn-SOD基因,WI-38,并鉴定了其特点。为了进一步阐明肿瘤细胞中人类Mn-SOD表达的减少机制,寻找到了缺少5′端区域的缺陷型,其能改变Mn-SOD基因的转录。图7-7所示的序列分析显示出在5′端区域中的NF-κB,AP-1,ARE,AP-2和SP-1的推断的结合位点,这表明人类Mn-SOD基因的表达可能受一些调节因子控制(见图7-7)。这个推断的启动子区域包含带有SP-1和AP-2转录因子共识结合位点的GC盒。要么SP-1结合位点自身重叠(SP-1/SP-1),要么SP-2结合位点与其重叠(AP-2/SP-1)。
7.2.3.2 癌细胞中Mn-SOD基因5′端区域的特点
采用四对引物从5′端扩增重叠片段,从SV40转化的人肺成纤维细胞(VA13)、人类结肠恶性细胞(HT29)和人前髓白血病细胞(HL60)分离出Mn-SOD基因转录起始位点下游的70bp的片段(见图7-8)。从人类肿瘤细胞系的基因组DNA扩增出来的产物的大小和正常细胞系是相同的并且与期望的大小一致。这些结果表明在检测的肿瘤细胞中人类Mn-SOD基因5′端区域没有添加或缺失。PCR产物的两条链的详细的DNA序列分析表明在PCR-1、PCR-2和PCR-3扩增产物中没有突变。然而在HT29肿瘤细胞系PCR-4区域中鉴定出三种突变体,但是在VA13和HL60细胞系中却没有(见图7-8)。这些突变是:-102位C突变成T,-38位C突变成G以及-93位含11个G的区域插入一个A。这个区域的突变的重要性被怀疑,因为PCR-4区域包含多重SP-1蛋白结合位点,这是人类Mn-SOD基因转录所需要的。在结肠癌和非结肠癌细胞系中检测PCR-4产物(见表7-2),发现在检测的五个结肠癌细胞系中有三个能够检测到相同的三个突变体(见表7-3)。人体成胶质细胞瘤U87以及人体成纤维细胞系HT-1080也携带有三种突变体。然而在人体乳腺癌细胞系和其他肿瘤细胞系中没有检测到这些突变。
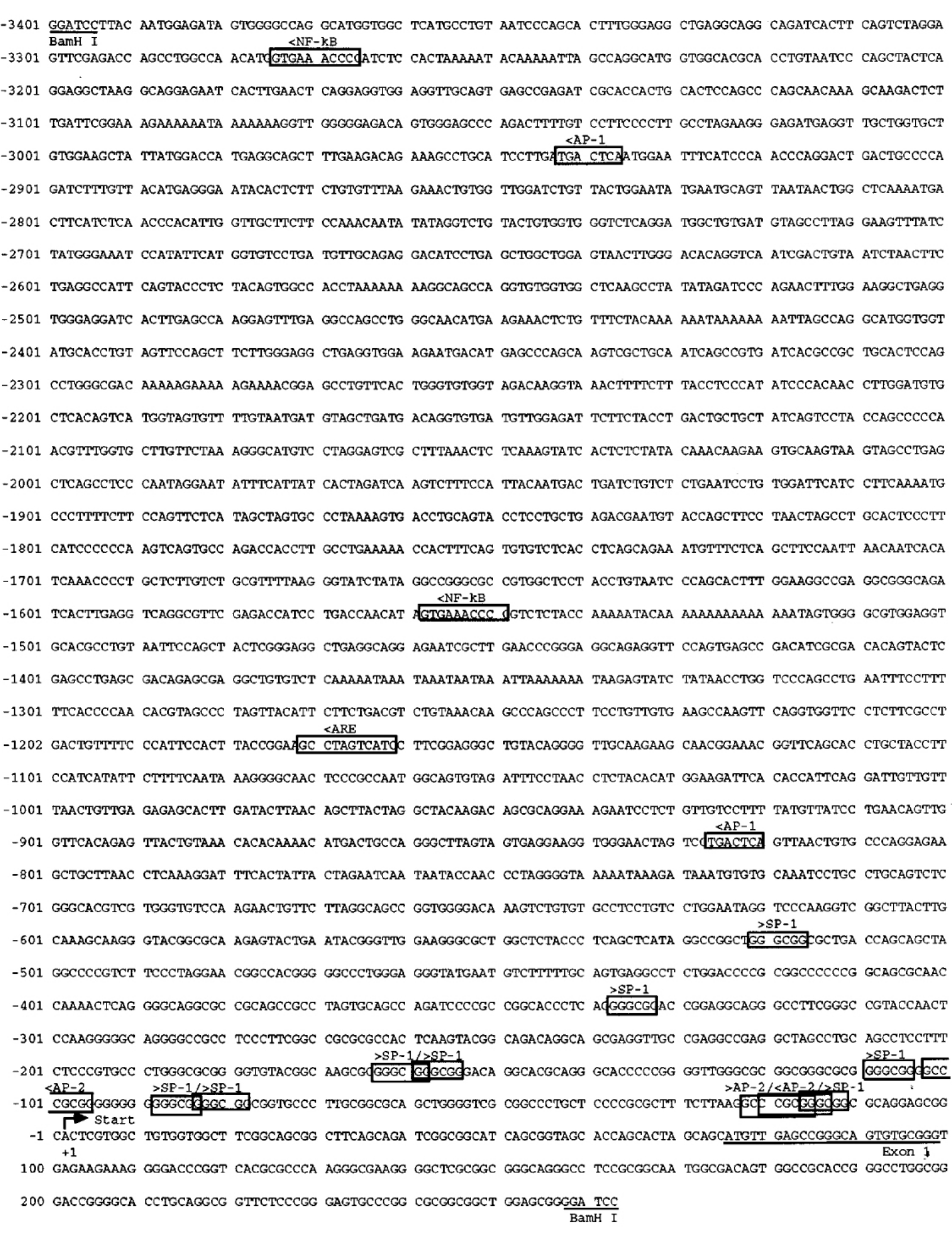
图7-7 人Mn-SOD基因的5′端区域的DNA序列
(摘自DK S.Clair,et al.Oncogene,1999)
转录起始位点是编号为+1的位点。序列编号是相对转录起始位点(+1)。框起来的是潜在的NF-κB,ARE,AP-1,SP-1和AP-2的转录调节结合位点。箭头表明检测出其中的共识序列
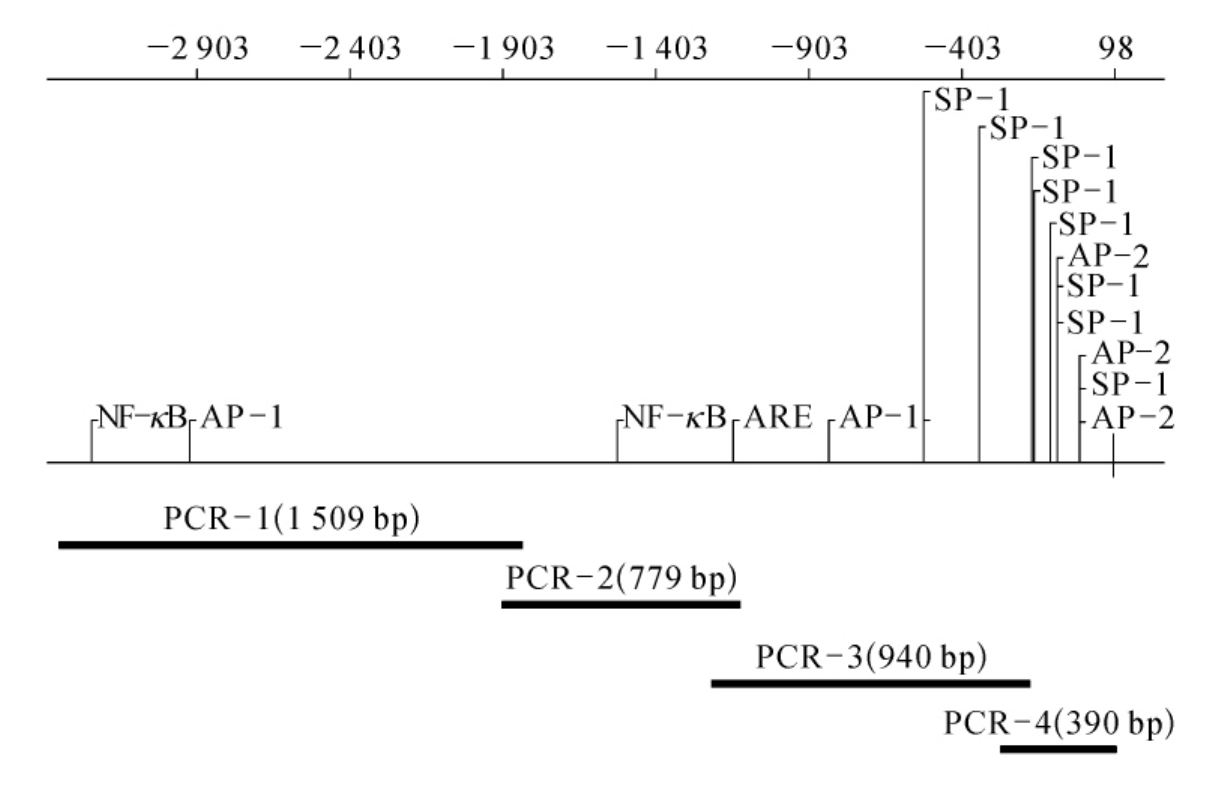
图7-8 转录因子在人类Mn-SOD基因5′端区域的结合位点图以及5′端区域的PCR扩增图
(摘自DK St.Clair,et al.Oncogene,1999)
相应的NF-κB,ARE,AP-1,SP-1和AP-2的转录因子的结合位点被标记出来(上)。肿瘤细胞系5′端区域扩增采用的四种PCR引物(下)
表7-2 本研究所用的细胞系
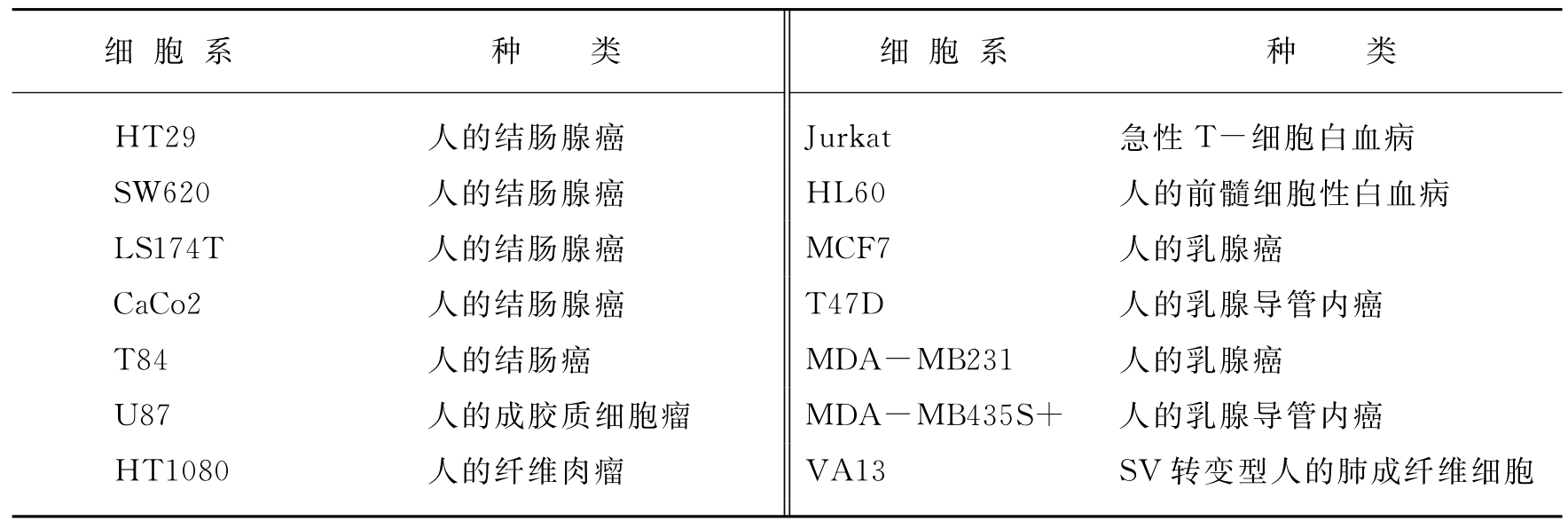
(摘自DK St.Clair,et al.Oncogene,1999)
表7-3 癌细胞中人的Mn-SOD启动子区域的突变(PCR-4)

(摘自DK St.Clair,et al.Oncogene,1999)
为了检测这些突变体是否是杂合的,肿瘤细胞系对应的PCR-4片段采用循环测序法进行直接测序。结果揭示了在三种突变位点有杂合现象(见图7-9,图7-10)。
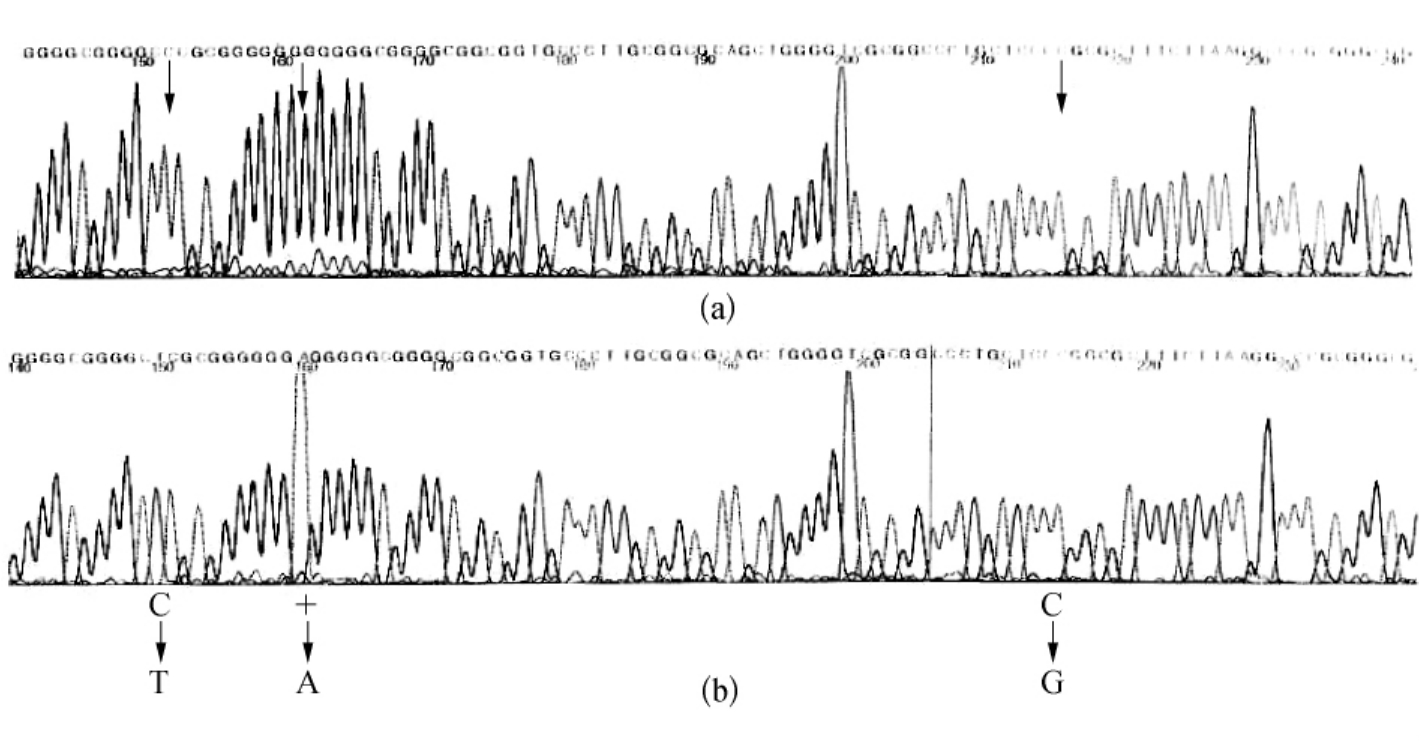
图7-9 肿瘤细胞系的PCR-4的产物的DNA序列分析
(摘自DK St.Clair,et al.Oncogene,1999)
(a)从分离出的野生型的序列WI38;(b)五种肿瘤细胞在特定位点的突变。在-102位点C突变成T,在-38位C突变成G以及在-93位插入一个A
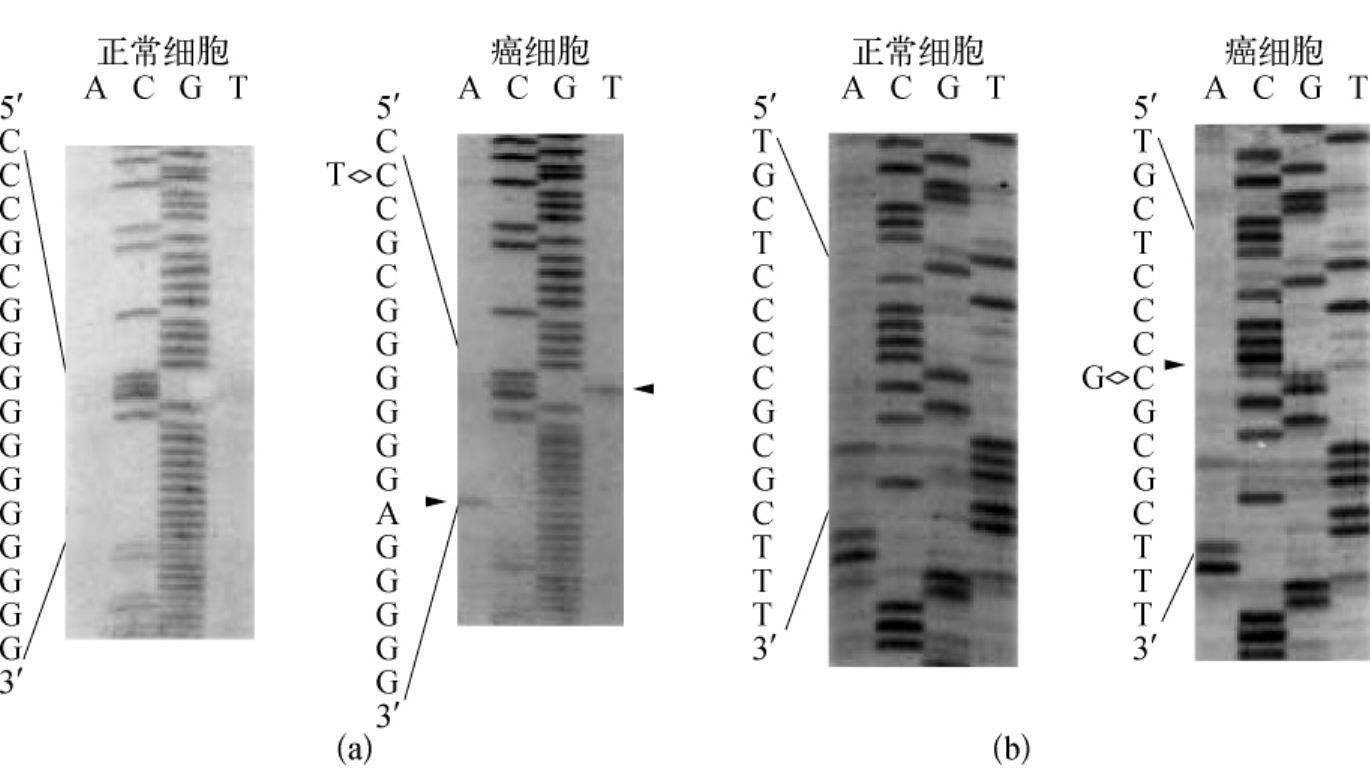
图7-10 在肿瘤细胞系中每个突变位点上用PCR产物直接测序法对杂合性的测定
(摘自DK St.Clair,et al.Oncogene,1999)
(a)指示在-102处C转换成T,在-93处插入一个A;(b)指示在-38处C到G的颠换。箭头所示的是突变位置。
电脑分析预测突变会改变限制性内切核酸酶的识别位点。在-93位插入A以及在-38位C突变成G将分别地创造一个新的MnlⅠ(CCTCN7)和一个AhaⅠ(CCSGG)限制性消化位点。然而,在-102位点C突变成T将导致ApaⅠ(GGGCCC)限制性消化位点的缺失。为了证实这些预测,克隆的片段(从-210到+24的启动子片段)用ApaⅠ进行消化,然后进行琼脂糖凝胶电泳。正常片段经ApaⅠ消化后产生两个条带(138bp和108bp)。来自肿瘤细胞的突变片段由于缺少了ApaⅠ片段只产生一个条带(246bp),如图7-11所示。
7.2.3.3 突变启动子的转录活性的降低
为了证明在人类Mn-SOD启动子的突变是否影响Mn-SOD基因的转录,从正常和癌细胞系中获得的PCR扩增的启动子片段(-154到24),带有或不带有三种突变,将其亚克隆进入基础pGL3和增强pGL3载体。用合成质粒瞬时转染VA13细胞,采用荧光素酶报告基因的表达来检验启动子活性,并以共转染的正常的荧光素酶的转染效率作为对照。正常的荧光素酶报告活性表明启动子的突变显著降低了启动子的活性(图7-12)。与正常启动子的活性相比,突变启动子在构建的pGL3质粒中活性下降了50%多,在构建的pGL3增强子中活性降低了90%。多次独立的转染得到了相同的结果。
7.2.3.4 突变启动子中转录因子结合方式的改变
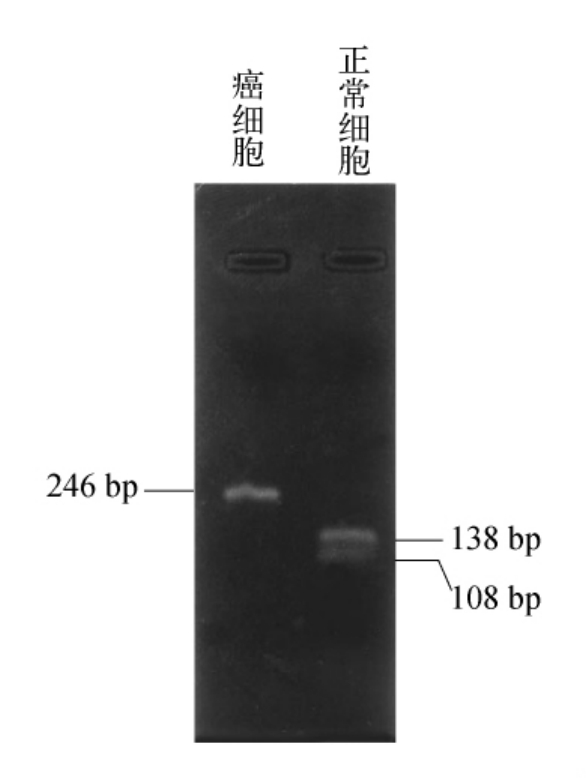
图7-11 启动子区限制性内切酶片段长度多态性分析
(摘自DK St.Clair,et al.Oncogene,1999)
-102处C到T的突变导致了肿瘤细胞系中ApaⅠ位点的缺失。ApaⅠ位点的突变产生了一个246bp的未切片段,ApaⅠ消化分成两个较小的片段(138bp和108bp)。
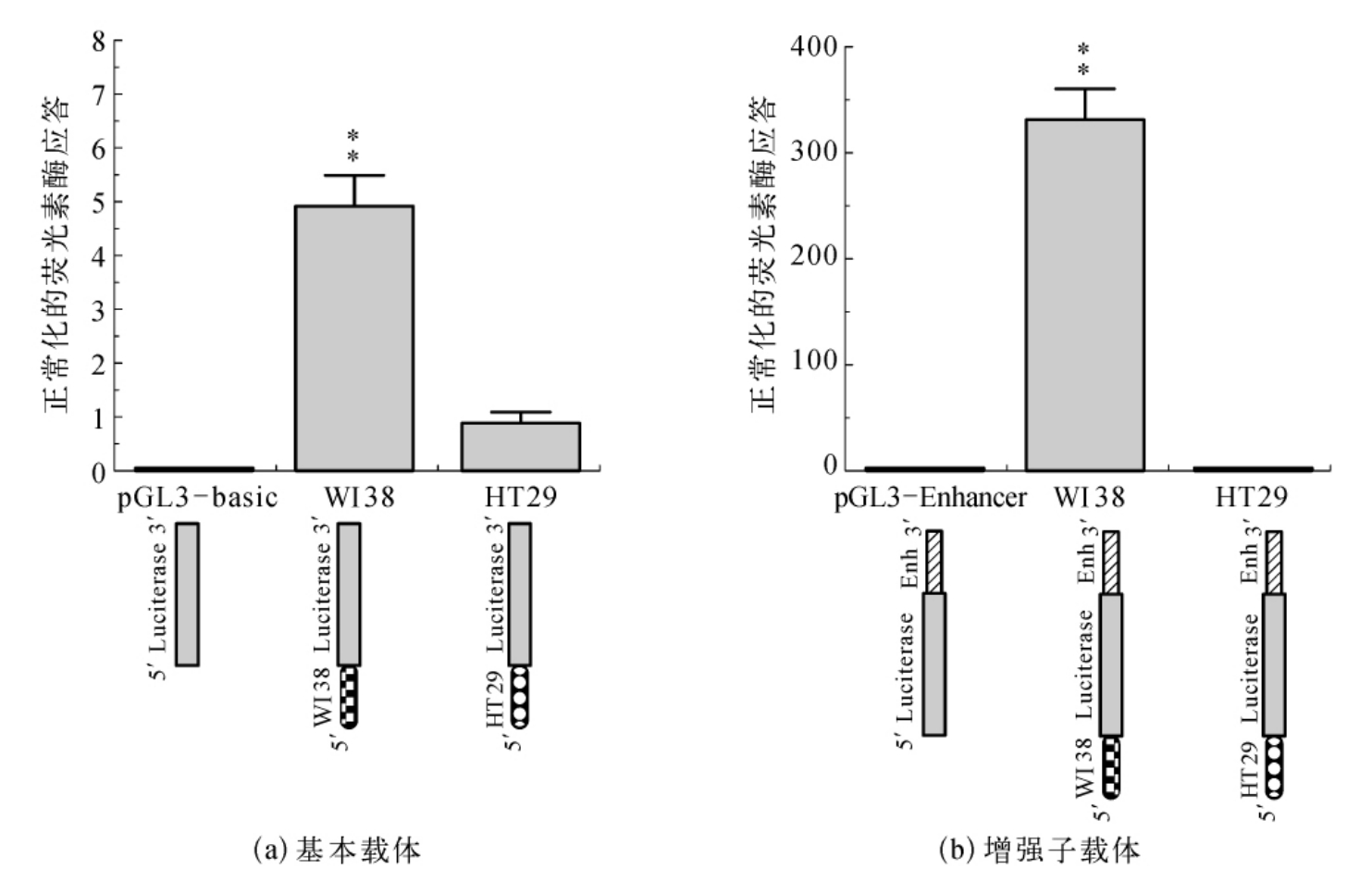
图7-12 人Mn-SOD启动子突变对基因表达的影响
(摘自DK St.Clair,et al.Oncogene,1999)
人类成纤维细胞被转染,使用了(a)pGL3基本媒介中克隆的质粒,它包含了人的Mn-SOD启动子和萤火素酶报告基因;(b)pGL3E媒介中的克隆质粒,而pGL3E是通过在荧光素酶基因的3′端侧翼区添加SV40强化因子对pGL3基本媒介进行修饰所获得。通过共转染的rellina荧光素酶对转染率变化的更正来使得活性标准化。转录活性中重要的差别看作是正常的启动子(P<0.01)。
用计算机在转录因子数据库中进行搜索,鉴定了在人类Mn-SOD基因启动子中有多个SP-1和AP-2转录因子的一致结合位点。我们的分析预测在突变启动子中AP-2而不是SP-1的结合方式改变。在-38位C和G的颠倒创造了一个新的AP-2结合位点以及在-102位点C和T的转换消除了一个AP-2结合位点。
为了确定突变启动子中转录因子结合位点的预期改变,在体外用纯化了的并带有放射性标记启动子片段(-210~+24)的SP-1和AP-2蛋白进行了DNase I印迹分析。在多结合位点处对该片段内的SP-1和AP-2进行印迹法观察(图7-13)。预测靠近转录起始位点处的SP-1位点似乎很微弱,探测不到。在正常启动子片段内,观察到五个强保护区域,它们与AP-2蛋白一道分别对应于位点1,2,3,5和6。一些AP-2结合位点也与SP-1结合位点相重叠(SP-1/AP-2)。当AP-2蛋白与突变启动子片段一起使用时,观察到另外一个AP-2保护位点(图7-13中的4区),这是与计算机搜索所得相一致的。然而,印迹法并没有表明-102处的C转变成T会使得AP-2结合位点的消失。并没发现SP-1结合构型的改变。
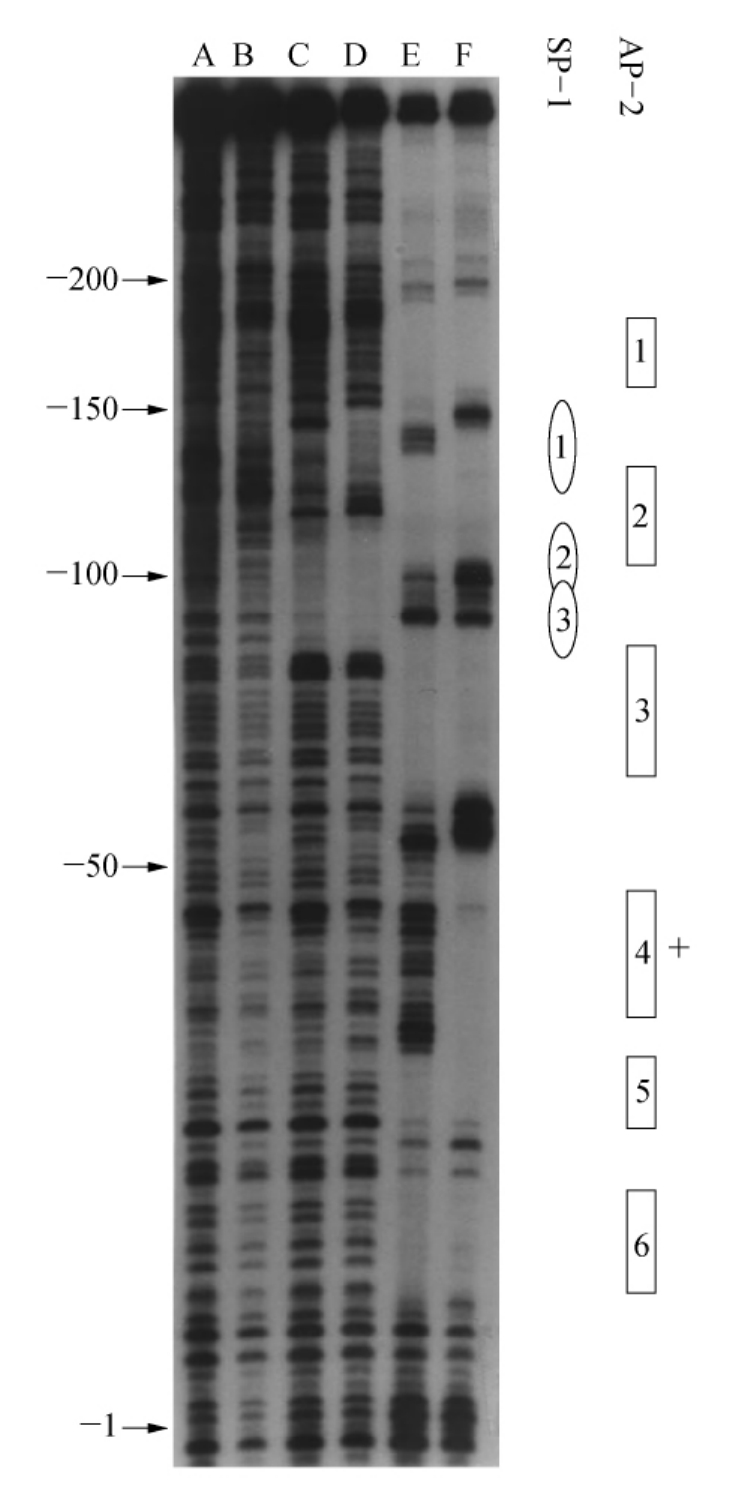
图7-13 用SP-1和AP-2蛋白对人Mn-SOD启动子的胰脱氧核糖核苷酸印迹法分析
(摘自DK St.Clair,et al.Oncogene,1999)
A—P7(一个正常的启动子片段,-210到+24)无蛋白控制;B—HT29(来自于HT29肿瘤细胞系中对应的片段)无蛋白控制;C—P7和SP-1蛋白;D—HT27和SP-1蛋白;E—P7和AP-2蛋白;F—HT29和AP-2蛋白。SP-1和AP-2的结合模式在右侧给出。(+)表示仅在突变启动子中发现的一个附加AP-2。
现已提出把Mn-SOD作为一种新的肿瘤抑制基因,因为在许多肿瘤和变异细胞之中Mn-SOD的活性是下降的,并且各种肿瘤中Mn-SOD的过度表达阻止了(细胞的)肿瘤化。以前的观察表明人肿瘤细胞中Mn-SOD活性降低的水平都是由于基因转录的缺陷造成的,因为处于稳定态的Mn-SOD mRNA浓度在肿瘤细胞中是减少的,并不伴有mRNA的稳定性的改变。在目前的研究中发现14个肿瘤细胞系,其中有5个被检测出携带共同的突变,突变位于Mn-SOD基因的启动子区。虽然使用细胞系进行研究有一个普遍的问题,这个问题在与肿瘤表型起源无关的自然突变的培养中可能发生,但这不太可能作为发现人Mn-SOD基因三个突变的案例,其理由如下:①这些突变的发生是把三个突变作为一组,均位于五个细胞系中相同的位点上;②五个人结肠癌细胞系中的三个都有这些突变的存在,而被测的四个人乳腺肿瘤细胞系中没有一个存在这些突变)。综合来说,这些结果表明在人Mn-SOD基因启动子区的突变可能在结肠癌中是显性的。
不论是在增强或是在非增强条件下,由肿瘤细胞中的突变启动子所启动的转化活性都极大地下降。这个结果说明,在某些肿瘤细胞里Mn-SOD活性的降低是由于(至少部分)该基因启动子的缺损。如表7-2所示,在所有被测的肿瘤细胞系中都没探测到突变。事实上,在VA13细胞中没有发现突变。针对肿瘤细胞中Mn-SOD的低量表达,之所以没有突变可能由于转录激活子和(或)阻遏物的存在。事实上,AP-2和SP-1在启动子区域内存在多个同序列位点,这表明转录因子AP-2可能与SP-1在调控Mn-SOD表达方面有着某些联系。已经证明在SV40转化细胞中,SV40-T抗原抑制了AP-2转录物激活的活性。因此,虽然继续对其进行研究,有可能是SV40-T抗原对AP-2活性的调节在VA13细胞中的Mn-SOD基因表达方面起着作用。
先前报道人Mn-SOD基因是一个单拷贝基因,它是由五个外显子组成,但被四个具有典型剪切位点的内含子所间隔。在小白鼠、牛、大鼠以及人的Mn-SOD启动子中富含GC(序列),具有保守的特点。哺乳动物富含GC的基因启动子对SP-1具有多个结合位点,并且位于5′端上游区的AP-2对于启动子的活性是十分必要的。目前的研究来看,从-210到+20这个片段包含了一个启动子区域,它与启动基因转录相配合。在五个肿瘤细胞重复片段中发现了这种启动子,启动子中的突变都是位于GC框,这表明在这些肿瘤细胞中Mn-SOD活性的表达可能被SP-1和AP-2相互作用所调节。
SP-1在所有的哺乳动物细胞中是普遍存在的转录因子。SP-1应答启动子通常在临近转录起始位点区含有多样的GC框。SP-1与GC框启动子元件相结合并通过RNA聚合酶Ⅱ选择性地激活mRNA的合成。据报道一些彼此接近的调节基因用具有高频率的G、C残基来保护启动子,这暗示了SP-1在这些基因的转录过程中起了极为关键的作用。然而,SP-1的亲和力和转录特性能够通过与其他辅助因子相互作用而改变。SP-1识别序列通常被发现存在于其他转录因子结合位点附近,例如CTF/NF-1,AP-1,NF-κB,C/EBP和AP-2,这一点表明这些因子可能通过相互之间的结合来调控转录。有趣的是,AP-2结合位点几乎在所有SP-1敏感启动子中都探测到了,并发现它涉及了基因的表达控制。一些研究报道:AP-2在组织特异性研究中对增强转录起着积极的作用。然而,据Getman等人报道AP-2能够作为人或小鼠的一种阻遏物对乙酰胆碱酯酶起作用,因为AP-2能够对SP-1竞争性地与结合位点结合,该位点处于启动子区的一个限制性片段内。研究表明,人类肿瘤细胞中Mn-SOD启动子的突变会导致AP-2结合构象的改变,而不是SP-1的改变。人的SP-1激活许多真核启动子的转录。SP-1的活化作用依赖于负责DNA结合的3个锌指结构以及至少一个富含谷氨酰胺区域(共2个),而谷氨酰胺富集区对转录激活是必须的。位于短链DNA片段中的早期临近位点里的SP-1蛋白,其相互作用使之形成了一些额外的结构,例如DNA环,DNA转折。人类Mn-SOD启动子是由多重SP-1和位于200bp DNA片段的AP-2结合位点组成的。多样SP-1和AP-2蛋白与其识别位点的结合可能会产生一个特殊的结构,该结构对其他调节蛋白的协同激活作用以及对该基因起协同转录激活作用的RNA聚合酶Ⅱ来说都是必需的。这启发我们去推测可能Mn-SOD启动子的突变改变了激活因子的结合方式,也可能干扰了DNA蛋白的相互作用并由此导致了这个特殊结构的破坏,抑或是改变了特异性结构,使得在起始复合物中抑制了蛋白质之间的相互作用。
现已证实细胞和病毒的基因都有形成二级结构的潜在性,尤其在富含GC的启动子中。比如,位于人的巨细胞病毒中的IE基因启动子被推测通过18个不成对核苷酸外部序列形成了一个十字结构。启动子中的这个二级结构与IE基因的高水平转录有关。假定的DNA成环结构是能够通过人类Mn-SOD启动子中的外部序列形成的(如图7-14所示)。在这个研究中,11个未配对的鸟嘌呤环可能为转录激活提供了一个结合区域。这个环状结构包括了基因基础转录所必需的三个SP-1和AP-2结合位点,该区域的完全缺失导致了90%以上的转录活性的降低。位于该区域中的三个突变,其中两个阻碍二级结构的形成,尤其是在-102位处的C转变为T可能导致推定是环状结构的改变。
总之,目前的研究是第一个去证明肿瘤细胞中Mn-SOD活性降低的程度部分是由于基因启动子区域的缺损。启动子区的突变使得DNA结合AP-2转录因子的形式发生改变。转录因子结合形式的调节可能会在DNA与蛋白质或者转录起始复合体中的蛋白质与蛋白质之间产生序列特异性作用的改变。这些突变并不发生在所有被检测的肿瘤细胞系中,这一点表明在某些肿瘤细胞中还存在着其他潜在的机制使得Mn-SOD的活性降低,人们当前也正对此进行研究。
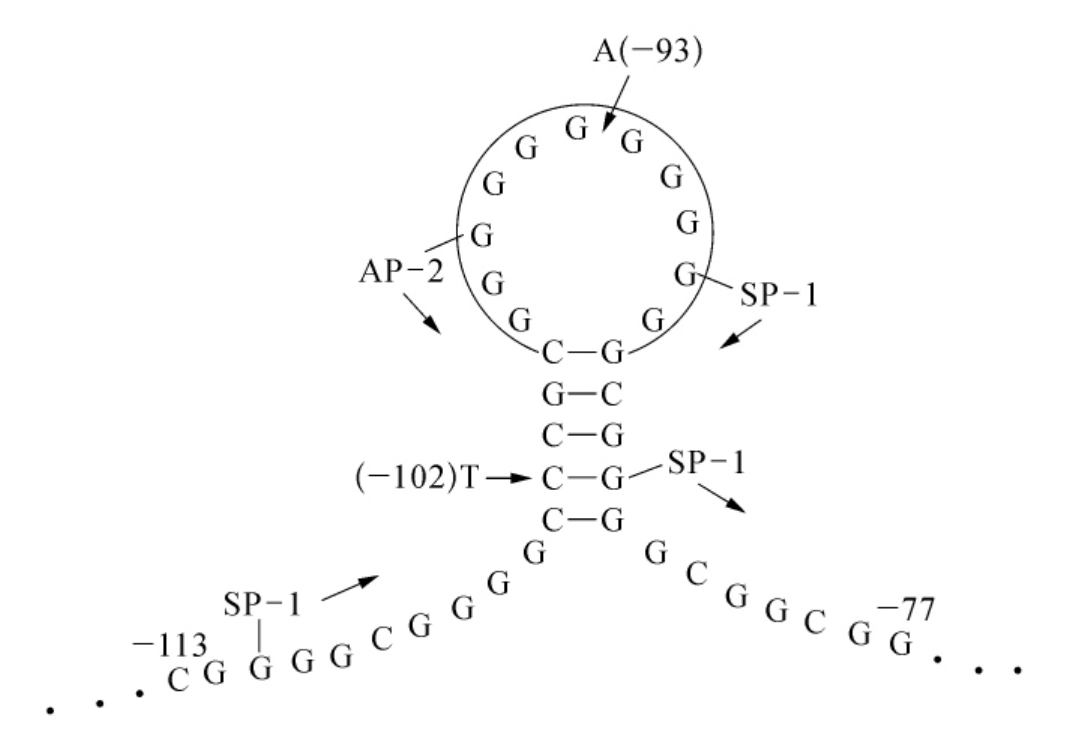
图7-14 在人Mn-SOD启动子中带有11-鸟嘌呤未成对的环的假定十字结构
(摘自DK St.Clair,et al.Oncogene,1999)
三个SP-1和一个AP-2结合位点位于DNA成环结构当中,并都被标记。有些位点也被指出,这些位点的突变可能是提出的结构断裂。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。









